











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCTION
The high prevalence of epilepsy affects more than 70 million people worldwide1. Thus, different anti-seizure medications (ASMs) - each with its molecular mechanism - have been developed since the 20th century. The International League Against Epilepsy (ILAE) defines epilepsy as a brain disease with any of the following characteristics: (1) two unprovoked seizures separated by 24 h; (2) an unprovoked crisis with a risk of recurrence within 10 years similar to the risk of recurrence given by having two induced seizures (60%); and (3) a diagnosed epileptic syndrome2. When patients have been diagnosed, the first line of control is the prescription of ASMs3. However, on many occasions, these drugs' use can generate alterations in their absorption, metabolism, and degradation pathway of the ASMs.
Seizures caused by abnormal, excessive, or concurrent electrical activity in the brain are known as epileptic seizures. Neuronal hyperexcitability in epilepsy is caused by an imbalance between excitatory and inhibitory ionic currents. As a result, most ASMs target Na+, Ca2+, K+, and Cl- ion channels, along with membrane receptors: primarily glutamate and gamma-aminobutyric acid (GABA)1. Nevertheless, ASMs controlled seizures are not utterly safe for the body. These drugs are known to have serious side effects (Table 1). As a result, it is critical to evaluate the patient's clinical history and their response to treatment before initiating a pharmacological treatment to minimize the possible adverse effects. In some cases, the use of ASMs is debatable4. For example, the pharmacological treatment for benign epilepsy with centrotemporal spikes appears to be unnecessary if it is considered to come from a benign origin4. ASMs resistance affects up to one-third of patients5, posing a significant challenge in neurology to discover safer and more effective ASMs.
Table 1. Pharmacodynamics and adverse effects of anti-seizure medications
| Antiepileptic name | Mechanism of action | Adverse effects/toxicity |
|---|---|---|
| Acetazolamide | Inhibition of the carbonic anhydrase | Metabolic acidosis, nephrolithiasis, nausea, vomiting, paresthesia, and metallic taste |
| Benzodiazepines | Increase the frequency of Cl-channel opening of GABA A receptor | Hypotension, respiratory depression, nystagmus, apnea, drowsiness, ataxia, sedation, and cardiac arrest |
| Brivaracetam | Binding to the SV2A protein | Dizziness, drowsiness, and flu symptoms |
| Carbamazepine | Reduction of neuronal action potential, inhibition of NMDA-activated ion current, blocking of adenosine receptor | SIADH, fatigue, diplopia, ataxia, leukopenia, and skin rash |
| Ethosuximide | Antagonism of the postsynaptic T-type voltage-gated calcium channel | Drowsiness, insomnia, headache, ataxia, nausea, vomiting, diarrhea, anorexia, and hiccups |
| Fenfluramine | Increased extracellular serotonin levels, serotonin reuptake inhibition | Anorexia, drowsiness, ataxia, sedation, serotonin syndrome, pulmonary arterial hypertension, and valvular heart disease |
| Gabapentin | Inhibition of the α2δ calcium channel subunit | Dizziness, ataxia, edema, nystagmus, weight gain, and respiratory depression |
| Lacosamide | Slow inactivation of voltage-gated sodium channels | Drowsiness, dizziness, ataxia, diplopia, nystagmus, tremor, and headache |
| Lamotrigine | Sodium channel blocker, decrease extracellular glutamate and aspartate | Skin rash, fatigue, drowsiness, dizziness, diplopia, leukopenia, and insomnia |
| Levetiracetam | Binding to the SV2A protein, inhibition of presynaptic calcium channels | Drowsiness, dizziness, emotional lability, and headache |
| Oxcarbazepine | Block of voltage-gated sodium and calcium channels | Headache, dizziness, drowsiness, diplopia, nausea, dyspepsia, and hyponatremia |
| Phenobarbital (Barbiturates) | Increase the time of Cl-channel opening of GABA A receptor | Dizziness, nystagmus, ataxia, bradycardia, bradypnea, hypothermia, and hypotension |
| Phenytoin | Block of voltage-gated sodium channels | Nystagmus, diplopia, sedation, ataxia, hypotension, nausea, vomiting, folate deficiency, gingival enlargement, hypertrichosis, drug-induced lupus, and anorexia |
| Piracetam | AMPA receptor modulator, improves acetylcholine function | Insomnia, irritability, headache, tremor, weight gain, agitation, and bleeding |
| Pregabalin | Inhibition of the α2δ calcium channel subunit | Dizziness, drowsiness, weight gain, blurred vision, diplopia, edema, dysarthria, and myalgia |
| Tiagabine | GABA reuptake inhibition | Dizziness, asthenia, tremor, headache, memory impairment, and psychosis |
| Topiramate | Block of voltage-gated sodium and calcium channels, inhibition of the carbonic anhydrase, block of AMPA receptor | Dizziness, weight loss, nausea, diarrhea, rhinitis, asthenia, myopia, and glaucoma |
| Valproate | Block of voltage-gated sodium channels, increase GABA levels, inhibition of histone deacetylases | Drowsiness, anorexia, nausea, dizziness, alopecia, encephalopathy, hepatotoxicity, pancreatitis, and polycystic ovary syndrome |
| Vigabatrin | Inhibition of the GABA aminotransferase | Headache, insomnia, vision loss, diplopia, drowsiness, memory impairment, psychosis, and fatigue |
| Zonisamide | Block of sodium and T-type calcium channels, inhibition of the carbonic anhydrase | Dizziness, irritability, diplopia, memory impairment, depression, insomnia, paresthesia, and metabolic acidosis |
GABA: gamma-aminobutyric acid; SV2A: synaptic vesicle protein 2A.
Some medications may predispose patients to epileptic seizures as a side effect of their therapeutic use6. This would add another pathway of neurotoxicity in both epileptic and non-epileptic people. In some cases, these epileptogenic pathways have been exploited as experimental models of epilepsy, which has aided in new treatment findings. Similarly, exposure to certain toxins can cause epileptic seizures. In this regard, certain toxins have been used as effective pharmacological compounds as experimental models of epilepsy. This review aims to analyze the molecular and genetic mechanisms of neurotoxicity involved in epilepsy, both damages caused by epileptic seizures and the therapeutic administration of ASMs targeting the central nervous system (CNS) neurotoxicity in epileptic seizures induced by other drugs and chemicals is reviewed.
MOLECULAR MECHANISMS OF ANTIEPILEPTIC DRUGS
To understand the ASMs molecular mechanisms of neurotoxicity, it is important to remember that these drugs act primarily by either inhibiting or exciting ion channels (Fig. 1). The resulting modification of signaling pathways will be crucial in triggering a range of adverse effects as described below.
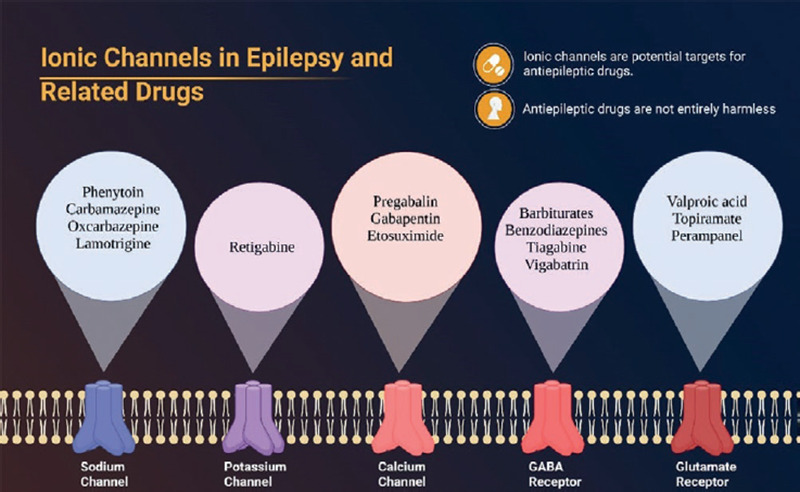
Figure 1. Ionic channels involved in epilepsy's pathogenesis and related drugs that act over them. Notice that some channels may be excitatory or inhibitory.
Sodium channels
Gated-dependent sodium channels are heteromeric complexes, composed of α and one or two β subunits encoded by SCN1A and SCN1B, respectively. SCN1A, SCN2A, SCN3A, and SCN8A genes, code for NaV1.1, NaV1.2, NaV1.3, and NaV1.6 channel subtypes, the primary sodium channels in the CNS. The α subunit is made up of four homologous but distinct domains (DI-DIV), and each of them contains six transmembrane segments (S1-S6). A central pore is produced between the four domains, in which the segments S5, S6, and the connecting pore-loops (P-loops) form the channel; while segments S1-S4 form the voltage-sensing domain7. SCN2A, SCN3A, and SCN8A genes are expressed mainly in excitatory neurons, whereas inhibitory interneurons express predominantly SCN1A and a low expression of SCN8A8. The expression - as well as the function of each channel - varies despite the similarity that exists between each of them; therefore, the presence of mutations or polymorphisms results in the manifestation of different seizure patterns and syndromes. Most of the mutations are found in the SCN1 gene. Mutation in SCN1A (Nav1.1) and SCN2A (Nav1.2) causes several subtypes of dominant idiopathic-generalized epilepsy (IGE)9. Particularly, SCN1A mutations are responsible for some types of epilepsy as Dravet syndrome and generalized epilepsy with febrile seizures (FS) plus. Some ASMs act on sodium channels, using their two states (closed and open state) to trigger their molecular mechanism10. The channel remains closed during the resting membrane potential, then quickly opens after depolarization and ultimately shuts to an inactive state. Depolarization is the physiological underpinning of epileptogenesis and the optimal target for ASMs since it is reliant on the sodium current (INa)11. INa blockers stabilize deactivated Na+ channels include phenytoin (PHT), carbamazepine, oxcarbazepine, and lamotrigine. Lacosamide (LCM) and eslicarbazepine are sodium channel blockers with a delayed inactivation rate12. The drug rufinamide has important adverse effects that occur in up to 60% of patients, including headaches, vomiting, dizziness, vertigo, drowsiness, and anorexia among others. These effects may be transient and have been reported to improve over time13.
Potassium channels
Voltage-gated potassium (Kv) channels are integral membrane proteins, belonging to the largest ion channel family in the human, encoded by 40 genes, and divided into 12 subfamilies (Kv1-Kv12). They play an important role in the action potential generation and propagation. Besides, Kv channels regulate the threshold potential for firing, the duration of action potentials, and the firing rates. Kvs' physiological role is defined by their localization within neuronal compartments14. The Kv channel is a homotetramer composed of four identical alpha (α) subunits that generate a potassium ion-selective pore in cell membranes14. Each α subunit has six α-transmembrane domains (TMD) (S1-S6), a membrane-entering P-loop located between the S5 and S6 domains, and cytosolic N- and C-terminus. The pore is formed by the segments S5-P-S6, while the sequences S1-S4 are essential for voltage sensing and channel activation15. These types of channels increase their functional diversity by assembling with other types of auxiliary subunits (β-subunit, Kvβ), which by themselves do not have a functional activity; but alterations in them can affect Kv channel function and network excitability, leading to increased seizure susceptibility13-15.
Voltage-gated dependent potassium channels are also involved in several processes such as cellular proliferation, migration, neurotoxicity, neuroprotection, and neuroregulation. More than 300 KCNQ1, KCNQ2, and KCNQ3 mutations have been identified to cause epilepsy including epileptic encephalopathy (EE) and benign familial neonatal epilepsy16. Due to their repolarizing actions, these channels have been explored pharmacologically to treat epilepsy. One example is retigabine, an activator of potassium channels Kv 7.2/7.3.17. Retigabine is indicated for resistant partial seizure treatment, with or without secondary generalization. Patients receiving retigabine as treatment should be under strict medical supervision, because this drug is capable of producing changes in vision or retinal pigmentation18. If this adverse effect occurs, retigabine should be discontinued18.
Calcium channels
Calcium is essential for cell membrane excitability, because it is a cation that depolarizes the membrane, allowing neurotransmitters to be released from synaptic vesicles to the synapse. Voltage-gated calcium channels (CaV or VGCC) are transmembrane proteins that act to regulate cellular functions related to calcium. Depending on the sensitivity to membrane depolarization, two CaVs have been described: High voltage-activated (HVA) and low voltage-activated (LVA) calcium channels19. HVA channels are heteromultimeric proteins that consist of a Cavα1 subunit, plus ancillary Cavβ, and Cavα2δ subunits in a 1:1:1 ratio. LVA channels may consist of only the Cavα1 subunit. The Cavα1 subunit is the essential component of the pore channel, and ten variants have been identified and divided into three subfamilies according to their pharmacological properties and pore-opening kinetic: CaV1 and CaV2 for HVA and CaV3 for LVA. CaV1 contains four members, all of them giving rise to L-type currents. CaV2 contains three members, with P/Q-, N-, and R-type currents. Finally, CaV3 has three members, all producing type T currents20. CaVα1 subunits all have similar structures, separated into four domains (I-IV) each of which is composed of S1-S621.
Recently, 12 mutations in the CACNA1A gene, which codes for the transmembrane pore-forming subunit of CaV2.1, have been reported in 10 unrelated cases of epilepsy. The results showed that CACNA1A mutations were associated with pure epilepsy and the spectrum of epileptic phenotypes. In the same way, the Leu226Trp CACNA1A variant was associated with juvenile myoclonic epilepsy (JME), in three members of an Iranian family affected by IGE22.
Pregabalin and gabapentin block HVA calcium channels through the Cav2 subunit, inhibiting neurotransmitter release at the synapse and lowering neuronal excitability. Ethosuximide (ESM), which inhibits calcium (ICa+) currents in thalamic neurons through the T-type channel, is the medication of choice in the typical absence seizures23. At present, Ca2+ blockers are used to control epileptic seizures, within the ASMs that have their mechanism of action blocking presynaptic Ca2+ channels. In the L- and N-type calcium channel blockers group, we can find topiramate, lamotrigine, zonisamide, and valproate. On the other hand, drugs such as gabapentin and lamotrigine are known to be H current modulators. It is suggested that PHT inhibits Ca2+ flux. Among its most important adverse effects are blood dyscrasias, such as aplastic anemia, Stevens-Johnson syndrome, fever, mouth and throat pain, fatigue, and burning eyes23.
Chloride channels-GABA receptors
Chloride is an essential anion for maintaining electronegativity in neurons. Cl-‘s principal hyperpolarizing effects on neuronal transmission are mediated through the GABAA receptor (GABAAR). The most prevalent inhibitor neurotransmitter in the CNS is GABA. GABA can work through ionotropic (GABAAR) and metabotropic (GABABR) receptors. The first of these has been one of the most crucial therapeutic targets in ASMs24.
GABAAR are heteropentameric structures made up of five subunits. A total of 19 GABAA subunits have been described belonging to different families: α (1-6), β (1-3), γ (1-3), δ, ε, π, θ, and ρ (1-3). The receptor consists of two α subunits, two β subunits, and one g subunit, arranged around a central pore permeable to Cl-. Each subunit shares a common structure and is composed of four TMD (TMD: TM1-TM4)25. It has been proposed that the TM2 domain forms the channel pore, while the intracellular domain between TM3 and TM4 is the site that modulates receptor activity. Some agents act on GABAAR, an important receptor targeted by multiple ASMs. Benzodiazepines (BDZ), barbiturates, and loreclezole, actions on the GABAAR are the main or only known mechanism of anticonvulsant action. For topiramate, felbamate, retigabine, losigamone, and stiripentol, GABAAR modulation is one of several potential anticonvulsant mechanisms26. Barbiturates, such as phenobarbital, and BDZ, such as diazepam, are two types of medications commonly used as ASMs. Barbiturates, on the one hand, prolong the opening of the chloride channel in the GABAAR, whereas BDZs enhance the frequency with which it opens. Mutations in the GABAAR subunit are related to several types of idiopathic epilepsy, associated with FS27. Other mutations identified in patients with epilepsy are those found in the genes encoding the α1, α6, β2, β3, γ2, or δ subunits of GABAARs. Many of these mutations affect the normal traffic of the receptor generating partially or completely impairing their expression on the synaptic plasma membrane28.
In addition, GABA function has been aided by the development of new antiepileptic medicines. Tiagabine was created to inhibit presynaptic GABA uptake by neurons and glial cells. Vigabatrin is a non-competitive GABA transaminase inhibitor that enhances GABA's impact on synapses29.
Glutamate receptors
Glutamate is the primary excitatory amino acid in the CNS, and it is produced in neurons from L-glutamine. Glutamate affects neurons through ionotropic and metabotropic receptors. N-methyl-D-aspartate (NDMA), -amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA), and kainic acid (kainate) receptors are examples of ionotropic receptors30. Metabotropic receptors are made up of eight receptors (mGluR1-mGluR8) classified into three classes. Group I receptors (mGluR1 and mGluR5) are connected to Gq proteins at both the presynaptic and postsynaptic levels. In Groups II and III, receptors are primarily bound to Gi proteins at the presynaptic level31.
It has recently been described that those mutations of GRIN1, GRIN2A, GRIN2B, and GRIN2D genes, which encode the GluN1, GluN2A, GluN2B, and GluN2D subunits, respectively, have all been associated with different epileptic phenotypes, such as EE, Rolandic epilepsy, Landau-Kleffner syndrome, and pike-and-waves during slow-wave sleep syndrome. The most frequent mutations are located in the GRIN2A gene and are present in the most severe phenotypes of epilepsy32. Perampanel is a selective AMPA-receptor antagonist. On the other hand, felbamate is an NMDA-receptor antagonist, which reduces excitatory neurotransmission by preventing glutamate binding33.
Valproic acid (VPA) has numerous targets in its mode of action; it can improve GABA ergic activities, inhibit the excitatory action of glutamate in the NDMA receptor, block voltage-dependent sodium currents, and has additional anti-epileptogenic properties that are not fully understood34. Topiramate, a sulfamate-substituted monosaccharide with comparable mechanisms of action, has a beneficial antiepileptic effect by raising the seizure threshold35. Other molecular targets, such as levetiracetam and brivaracetam, are synaptic vesicle protein 2A (SV2A) modulator ligands that can limit neurotransmitter release36. Acetazolamide and other carbonic anhydrase inhibitors have also been authorized to treat epilepsy.
NEUROTOXICITY OF ANTI-SEIZURE MEDICATIONS
ASMs are the first-line treatment for practically every kind of epilepsy; nonetheless, these medications are not devoid of producing neurotoxicity or inducing epileptic episodes. While an ASMs may be the gold standard of therapy for one kind of epilepsy, it might exacerbate other types of epilepsy if overused. Wrong diagnoses, pharmacological tolerance, inverse pharmacodynamic effects, incorrect dosage by overdosing, and irregular medication consumption are all common reasons for this paradoxical result.
The medications of choice for childhood absence epilepsy include ESM and VPA, whereas carbamazepine, PHT, phenobarbital (at high dosages), and tiagabine might aggravate seizures23. In Lennox-Gastaut syndrome, the recommended medication is VPA, rufinamide, lamotrigine, or topiramate; however, gabapentin or BDZ aggravate seizures. JME can be treated with VPA, levetiracetam, and lamotrigine. However, it is exacerbated with carbamazepine (CBZ) and PHT37. PHT can also worsen myoclonus in symptomatic epilepsies. Infantile spasms traditionally treated with VPA and vigabatrin (or adrenocorticotropin in the setting of West syndrome) may be exacerbated by CBZ and BDZ38. The VPA is the ASM with the most therapeutic indications in epilepsy, due to the broad range of its mechanism of action; nevertheless, this suggests that enough side effects have emerged. Dose-dependent side effects include gastrointestinal abnormalities such as anorexia, nausea, and vomiting, which usually subside at low doses, there may be increased appetite with subsequent weight gain; the phenomenon of hypersensitivity is occasionally expressed by a skin rash; and up to 50% of patients may notice hair thinning and fragility, with alopecia occurring in some cases39. Menstrual irregularities and polycystic ovarian syndrome have been linked to VPA intake, according to case studies40. In terms of neurotoxicity, it should be noted that the appearance of distal fine tremor, while not significant enough to withdraw the drug. In addition, it should be noted that hyperammonemia encephalopathy may occur in severe cases, so VPA should not be administered in patients with liver disease or mitochondrial diseases41. Pseudoatrophy of the brain has been observed in several cases.
PHT is an antiarrhythmic medication. Chronic usage causes gingival hyperplasia, hirsutism, coarse facial features, hyperpigmentation, and acne, along with changes in bone metabolism and folate deficiencies at the systemic level42. Serum levels > 30 µg/mL are associated with vestibular system dysfunction, which manifests as nystagmus, ataxia, dysarthria, and, lastly, consciousness level abnormalities. Personality changes, peripheral neuropathy, and extrapyramidal symptoms have also been observed on a rarer basis. Chronic PHT treatment can result in permanent cerebellar atrophy43.
CBZ is a medication that has a similar structure to TCA. Despite being a highly effective treatment for focal onset seizures, it should be used with extreme caution in generalized seizures due to the paradoxical consequences. Most of its toxic effects are manifested at the CNS level and include somnolence, tiredness, dizziness, diplopia, ataxia, and asterixis. Other systemic side effects include secondary hyponatremia, SIADH, agranulocytosis, leukopenia, and decreased cardiac conduction44.
Phenobarbital (PHB) is a barbiturate having hypnotic and sedative characteristics. Its neurotoxic effects are dose-dependent and quite like those seen with other ASMs, including ataxia, tiredness, drowsiness, behavioral abnormalities, mood disorders (depression), libido issues, and sexual impotence. The above is traditionally related to Dupuytren contracture, along with folate, cobalamin, and thyroxine insufficiency45.
BDZ (diazepam, clonazepam, clobazam, midazolam, and lorazepam) has different pharmacological interactions based on their half-life. However, they share neurological dose-dependent side effects that can be mild, such as fatigue, dizziness, somnolence, ataxia, and irritability. At higher doses, it can cause life-threatening situations such as arterial hypotension and respiratory depression46.
Most of the other ASMs have the same neurological side effects as the medications listed above. Nonetheless, many significant side effects should be noted. Vigabatrin (VGB) promotes GABAergic cell malfunction in the retina cone cells, resulting in visual field changes and irreversible blindness. Suicidal thoughts may also be increased with vigabatrin47. When it comes to topiramate (TPM), almost half of the individuals using this treatment have paresthesia and weight loss. Verbal language impairments, on the other hand, are rather prevalent. Psychotic symptoms have been reported in individuals with a psychiatric history at high doses of TPM, perhaps due to the drug's suppression of the prefrontal cortex48. Although no therapy is completely unassailable for the body, the tolerance to new antiepileptics has increased the prescription for epilepsy treatments. Levetiracetam is one of these treatments, with modest and potentially reversible neurotoxic effects and reduced pharmacological interaction49. Under the right treatment regimens, both lamotrigine and LCM are typically well-tolerated.
EPIGENETIC TOXICITY DUE TO ANTI-SEIZURE MEDICATIONS
As mentioned above, ASMs are directed to different molecular targets to exert their effects as regulators of voltage-dependent ion channels or synaptic excitability. The above implies that these functions also consequently modify gene expression, functioning as epigenetic modifiers. Epigenetics refers to all those gene-dependent changes that are heritable but lack alterations in deoxyribonucleic acid (DNA) sequencing. The point of association between drugs and their gene effects is found in epigenetic markers (Fig. 2A and B). The modification of histone proteins, DNA methylation, and untranslated RNA functions are key markers50.
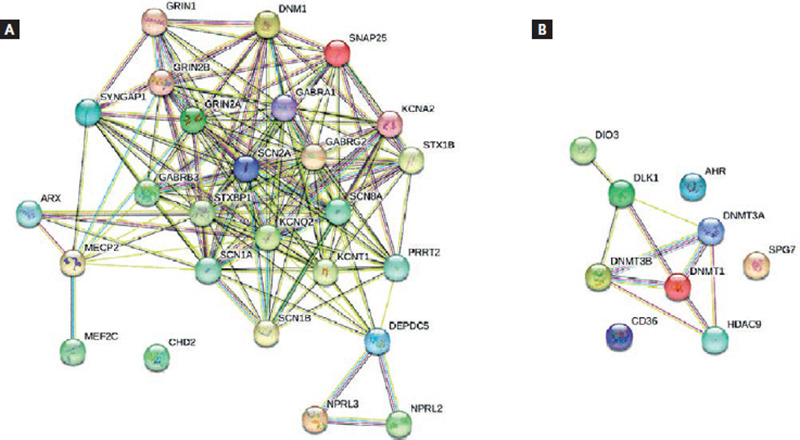
Figure 2. Interaction networks in epilepsy. Nodes represent proteins and edges represent protein-protein associations (i.e., proteins jointly contribute to a shared function). Filled nodes represent some 3D structure is known or predicted. (A) The most significant genes implicated in epilepsy. The genes mainly involved encode subunits of ion channels: sodium (SCN1A, SCN2A, SCN1B, and SCN8A), potassium (KCNQ2, KCNA2, and KCNT1), and GABA-chloride (GABRA1, GABRG2, and GABRB3). SCN2A stands out for its multiple associations with almost all the genes. Analysis: average node degree: 12.5, average local clustering coefficient: 0.792. (B) The most significant genes altered with antiepileptic treatment. It can be seen that the three genes with greater importance in these associations are DNMT3A, DNMT3B, and DNMT1. These genes encode DNA (cytosine-5)-methyltransferases. DNMT3A and DNMT3B methylate genome-wide de novo and establish methylation patterns during development. DNMT1 methylates hemimethylated DNA. Analysis: average node degree: 2.22, average local clustering coefficient: 0.556 (Retrieved from: https://string-db.org/cgi/network.pl).
Histone modification is primarily involved in transcription manipulation through acetylation. This process is regulated by groups of specific enzymes that cause metabolites to serve as gene activators or inhibitors, functioning as a balance between histone acetyltransferases and histone deacetylases. The latter is classified into four classes, and it has been shown that drugs such as VPA, CBZ, TPM, and LCM can inhibit them, mainly class I. An inhibition of this enzyme group provokes the accumulation of acetylated protein forms, regulating in turn cell proliferation and cell death51.
The DNA methylation process - the second epigenetic biomarker - is interfered with by de novo or maintenance methyltransferases (DNMTs), which are responsible for guiding a stable, inherited gene regulation essential in epigenetic regulation. These enzymes add a methyl group to DNA during or after DNA replication. Three groups of DNA methyltransferases (DNMT1, DNMT3a, and DNMT3b) have been identified in mammals. Drug exposure affects methylation, inducing a state of hypermethylation or hypomethylation in the promoter regions of genes. VPA is a known inhibitor of all three enzymatic forms of DNMTs, which influences neuronal migration and brain development52. Other drugs such as ESM have a potentiating effect by elevating DNMT1 and DNMT3a expression when administered chronically30.
Untranslated RNA lacks protein-coding potential on its own, but its incidence when expressing other genes is key in this type of epigenetic biomarker31. PHB has also been associated with alterations by this mechanism, specifically in abnormal expression at delta-like homolog 1 gene (Dlk1) and the iodothyronine deiodinase type III gene (Dio3) levels. The expressivity modifications in these genes are related to hepatocytes' hypertrophy with oncogenic capacity53. The intervention of ASMs at the three epigenetic mechanisms discussed above has negative health consequences, which the researchers call “epigenetic toxicity.” When we talk about genetic alterations, we immediately think of oncogenes. In this sense, ASM has been associated with carcinogenic promoters, for example, it has been seen that PHB by altering the expression of non-coding RNA by activating carcinogenic induces the transformation of hepatocytes into neoplastic cells54.
ASMs also have neurotoxic potential, as they have been linked to alterations in genes key to neuronal development in the fetal stage. The genes primarily affected are those that code for placental transporters and folate metabolism. Of all the drugs, the one with the highest teratogenic risk is VPA since studies have shown that its involvement in acetylation and histone methylation affects the neuroepithelium in rats. Mice with administration schedules of this drug have shown elevated levels of epigenetic markers recognized in this pathology such as miR-132 and miR-134-5p in their nerve tissue. Furthermore, autism spectrum disorder has even been associated with the use of VPA55. An inflammatory effect of genetic origin is another of the mechanisms of toxicity that has been found in epilepsy treatments. VPA decreases the methylation levels of the PPARγ, PPARα, CD36, and AHR genes, which causes an inhibition of beta-oxidation and an increase in the hepatic entry of fatty acids. The result will be an inflammatory chain that starts from the consequent hepatic steatosis. VPA is also a known drug-causing agent of pancreatitis, interfering with the proliferation of acinar cells and reprogramming their embryological configuration by inhibiting HDAC56. Another drug associated with activation of the inflammation cascade is CBZ, which can activate CD4+ T lymphocytes by epigenetic factors such as miR-18a and miR-155. This lymphocyte activation will favor immune hypersensitivity processes such as Stevens-Johnson syndrome57.
DRUG-INDUCED EPILEPTIC SEIZURES
The fact that certain drugs used for different purposes can trigger epileptic seizures has allowed to deepen the research both of epileptogenesis and adverse effects of ASMs, since some of these drugs are used as experimental models of epilepsy58. Penicillin administration is one of the chemical models of epilepsy most used in experiments59. The drugs that can cause epileptic seizures and their respective mechanisms of ictogenesis are described, which allows us to have more caution when selecting a drug for a patient with comorbidities related to epilepsy.
Antibiotics
Antibiotics, especially beta-lactams (penicillin, cephalosporins, monobactams, and carbapenems), are a class of medications that have been extensively explored for these effects. The GABAAR is the most prevalent toxicity target for these medicines (Fig. 3). Penicillin, cephalosporins, imipenem, and fluoroquinolones, among others, may function as direct antagonists of the GABAAR in the CNS in susceptible individuals. Isoniazid, on the other hand, an antitubercular drug, has two identified mechanisms: competitive inhibition of pyridoxine kinase and blockage of glutamate decarboxylase, in which both actions impede GABA production60.
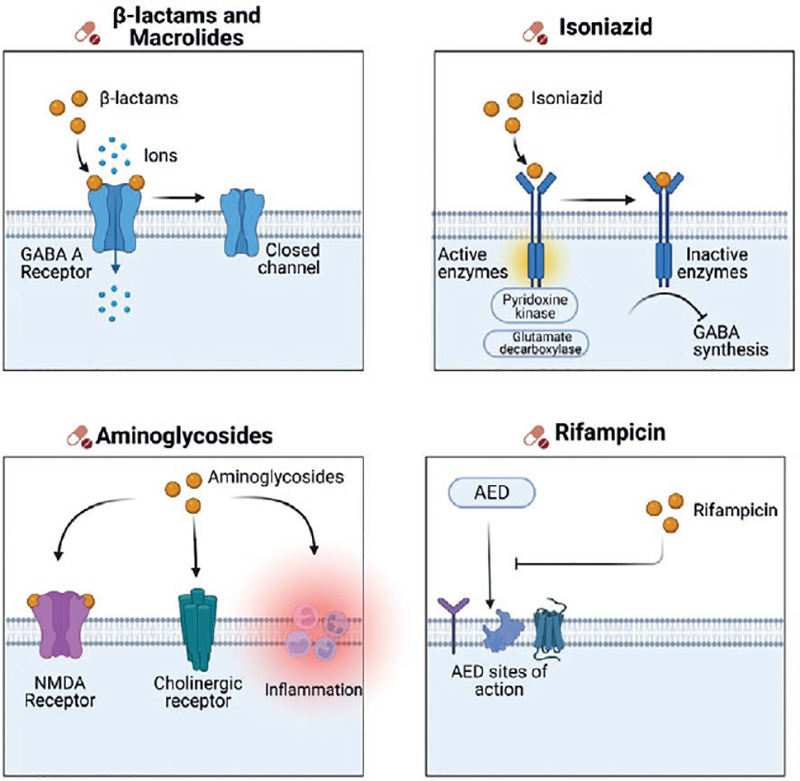
Figure 3. Drug-induced epileptic seizures. Beta-lactams and macrolides have effects on suppressing the activity of GABAΑ receptor. Isoniazid has two identified mechanisms: Competitive inhibition of pyridoxine kinase and blockage of glutamate decarboxylase, which both actions impede GABA production. Aminoglycosides may interact with NDMA receptors, cholinergic receptors, and may cause an inflammatory response in the axon. And lastly, rifampicin can lower blood levels of carbamazepine, valproic acid, ethosuximide, and phenytoin.
Macrolides may cause seizures, according to limited research. Clarithromycin, which is routinely used to treat upper respiratory tract infections, has been examined as a stimulant, specifically for the treatment of hypersomnia. Clarithromycin-induced reversible cellular hyperexcitability was reported by Bichler et al., and it is suggested that this action is mediated through the GABA receptor61. There have been reports of epileptic seizures induced by the therapeutic administration of metronidazole, polymyxin B, and colistin, but no convincing evidence on the action mechanism is available62.
Increased permeability of the blood-brain barrier in CNS disorders and prematurity, as well as impaired drug metabolism in kidney and liver failure are risk factors for developing drug-induced seizures60. Another notable neurotoxic impact of antibiotics is that of aminoglycosides, which produce ototoxicity, peripheral neuropathy, and possibly encephalopathy by activating NDMA receptors, interacting with cholinergic receptors, and causing an inflammatory response in the axon. Gentamicin is the primary cause of adverse symptoms63.
Furthermore, we must examine the pharmacological interaction under polypharmacy situations. For example, rifampicin can lower blood levels of CBZ, VPA, ESM, and PHT. On the other hand, trimethoprim-sulfamethoxazole enhances PHT toxicity60.
Stimulants and sympathomimetics
For several years, the role of noradrenaline in epileptic seizures has been demonstrated. Experimental models of epilepsy based on ligands of adrenergic receptors have been developed; therefore, drugs with adrenergic agonist action could be expected to cause epileptic seizures as side effects, as with phenylephrine and pseudoephedrine. A class of powerful CNS stimulant medications known as amphetamines is synthetic adrenergic agents. Methylphenidate, an amphetamine structural analog, is a medication licensed for the treatment of attention deficit hyperactivity disorder. It works by boosting dopamine and noradrenaline levels in the CSN by inhibiting monoamine carrier recapture, resulting in dangerous levels capable of inducing epileptic seizures64.
Certain psychopharmaceuticals can cause epileptic seizures through a similar mechanism, including monoamine oxidase inhibitors (isocarboxazid, phenelzine, and selegiline), selective inhibitors of serotonin reuptake (fluoxetine, sertraline, and paroxetine), serotonin-norepinephrine reuptake inhibitors (atomoxetine and desvenlafaxine), and tricyclic antidepressants (amitriptyline, imipramine, and nortriptyline). Bupropion is a dopamine and noradrenaline recapture inhibitor intended to help people quit smoking, according to Yan65. Despite being well-tolerated in his early clinical trials, it has subsequently been linked to seizures following an accidental or purposeful overdose. This dose-dependent impact is directly tied to the history of epilepsy. Nevertheless, the mechanism of action by which seizures occur has not yet been properly defined.
Other drugs associated with seizures
However, research on the capacity of non-steroidal anti-inflammatory drugs and opioids to cause epileptic seizures is yet inconclusive. Both acetaminophen and ibuprofen appear to be medicines that, at therapeutic levels, would not lower the seizure threshold. Due to adenosine A1-receptor antagonism, methylxanthines (theophylline and aminophylline) can produce seizures7. Antidiabetic medications, especially those with a strong hypoglycemia impact, might cause seizures. Seizures develop at blood glucose levels of 16-19 mg/100 mL according to experimental research. Furthermore, it has been reported, that there are spontaneous sporadic occurrences of epileptic seizures following the administration of anticancer medicines and immunosuppressants66.
CONCLUSION
As we have discussed, the evidence of the adverse effects prevalence due to antiepileptic drugs administration is significant. These findings are not surprising, since each individual response may vary depending on the molecular mechanisms involved with the epilepsy type, as well as the drug used. In this regard, epigenetic factors have a relevant function in the regulation of mechanisms involved in ASMs processing. Many ASMs generate epigenetic toxicity that can lead to gene dysregulation and thus to the possible cancer development by promoting neoplastic cells generation. In addition, the presence of genetic variants in the different ionic channels is relevant to defining the prognosis and type of treatment used. It is not surprising that the response to a drug can vary between individuals, who present the same phenotype. Hence, the importance of creating new strategies that mitigate the side effects of ASMs, considering the individual aspects of each patient and their comorbidities to develop personalized therapies.

