











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkThe virus
The causal agent of the COVID-19 pandemic is the Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) first described in Wuhan, China in December of 2019 (Lu et al., 2020; Zhu et al., 2020). Two other coronaviruses are highly pathogenic to humans. The Severe acute respiratory syndrome coronavirus (SARS-CoV) was described in China in 2002, and the Middle East respiratory syndrome coronavirus (MERS-CoV) was described in South Arabia in 2012 (Cui et al., 2019). Both SARS-CoV and SARS-CoV-2 originated in bats, in China, and adapted to infect humans (Cui et al., 2019; Cagliani et al., 2020; Lu et al., 2020).
Coronaviruses form spherical virions with a membrane envelope. The genome is single-stranded RNA (Cui et al., 2019). As in all RNA viruses, in coronaviruses sources of genetic variation include nucleotide insertions, deletions, substitutions and include RNA recombination. These events occur naturally during RNA replication (Sanjuán and Domingo-Calap, 2016). Genetic variation and selection favor accumulation of mutations in parts of the genome responsible for critical processes, such as host adaptation, vector transmission, entry into the cell, and suppression of antiviral defense (Obenauer et al., 2006; Nigam and Garcia-Ruiz, 2020).
At the population level, genetic variation and selection drive the formation of new strains and species (Lauring and Andino, 2010). This model supports the emergence of SARS-CoV and SARS-CoV-2 in bats followed by adaptation to humans (Cui et al., 2019; Cagliani et al., 2020; Lu et al., 2020). SARS-CoV never reached pandemic level. One of the differences is that SARS-CoV-2 is more readily transmissible than SARS-CoV. The genetic difference in transmissibility and pathogenicity maps to the spike protein S (Zhou et al., 2020).
The spike protein decorates the coronavirus virion and mediates entry into the cell to initiate infection (Li, 2016; Wrapp et al., 2020a). SARS-CoV-2 entry is mediated by the specific interaction between the spike protein S and cellular receptor angiotensin-converting enzyme 2 (ACE2) (Cai et al., 2020). Infected people develop neutralizing antibodies against the entire protein S and non-neutralizing antibodies against fractions or a subunit of protein S (Brochot et al., 2020; Cai et al., 2020). Accordingly, antibodies against the S protein are used as markers in diagnostic assays (Zhu et al., 2007; Li et al., 2008; Brochot et al., 2020). Other, coronavirus diagnostic protocols are based on the detection of viral RNA by RT-PCR, viral proteins, or antibodies developed against viral proteins (Brochot et al., 2020; Phan, 2020b; Zhu et al., 2020).
Vaccines against SARS-CoV-2 are being developed using multiple approaches, including attenuated or inactivated viruses, DNA, adenovirus-based and mRNA vaccines (Amanat and Krammer, 2020; Dearlove et al., 2020). In the United States of America, two mRNA vaccines based on protein S have been authorized for use by the Food and Drug Administration. The end goal of these vaccines is to block virus entry into the cell by activating the formation of antibodies against protein S. We recently showed that the cistron coding for protein S is the most variable in the genome of SARS-CoV-2 and in all species in the genus Betacoronavirus (LaTourrette et al., 2021), which includes SARS-CoV and MERS-CoV. The wide genetic diversity of their host has selected Betacoronavirus for hyper variation in protein S (LaTourrette et al., 2021).
The hyper variable nature of protein S has several biological functions. One is to maintain functionality and recognize a genetically diverse group of potential hosts, such as humans or bats (Zhai et al., 2020). Another is to trigger the formation of non-neutralizing antibodies that serve as decoys. Protein S variation may also escape neutralizing antibodies formed by natural infection or triggered by vaccines (Long et al., 2020; Walls et al., 2020). Accordingly, efficacy of vaccines, and reliability of antibody-based diagnostic test has potential to be affected by variation in protein S. Protein S variation also explains the occurrence of repeated SARS-CoV-2 infections (Tillett et al., 2020). Fundamental understanding of the coronavirus biology and genomic variation establish the basis for designing and deploying diagnostic tests, vaccines, and antiviral drugs.
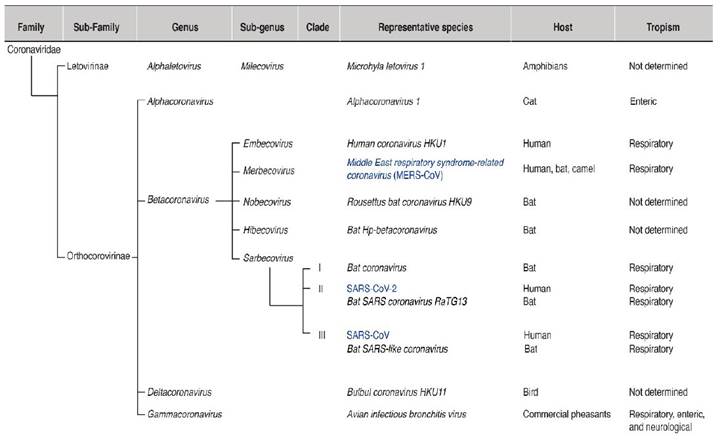
Coronavirus taxonomy
Coronaviruses belong to the order Nidovirales, the family Coronaviridae, the sub-family Orthcoronavirinae, and four genera (Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus) (Lu et al., 2020; Zhu et al., 2020) (Figure 1). Alphacoronaviruses infect mammals. Betacoronaviruses mainly infect bats and humans. Gammacoronaviruses and Deltacoronaviruses infect birds, and some species infect mammals. The genus Betacoronavirus is divided into five sub-genera (Figure 1): Embevorirus, Merbecovirus, Nobecovirus, Hibecovirus, and Sarbecovirus (Lu et al., 2020; Zhu et al., 2020). The sub-genus Sarbecovirus contains species that infect only bats or humans and includes SARS-CoV and SARS-CoV-2. Another human coronavirus, MERS-CoV, belongs to the sub-genus Merbecovirus. The sub-genera Nobecovirus, Hibecovirus are integrated by species that infect bats (Figure 1).
Coronavirus genome organization
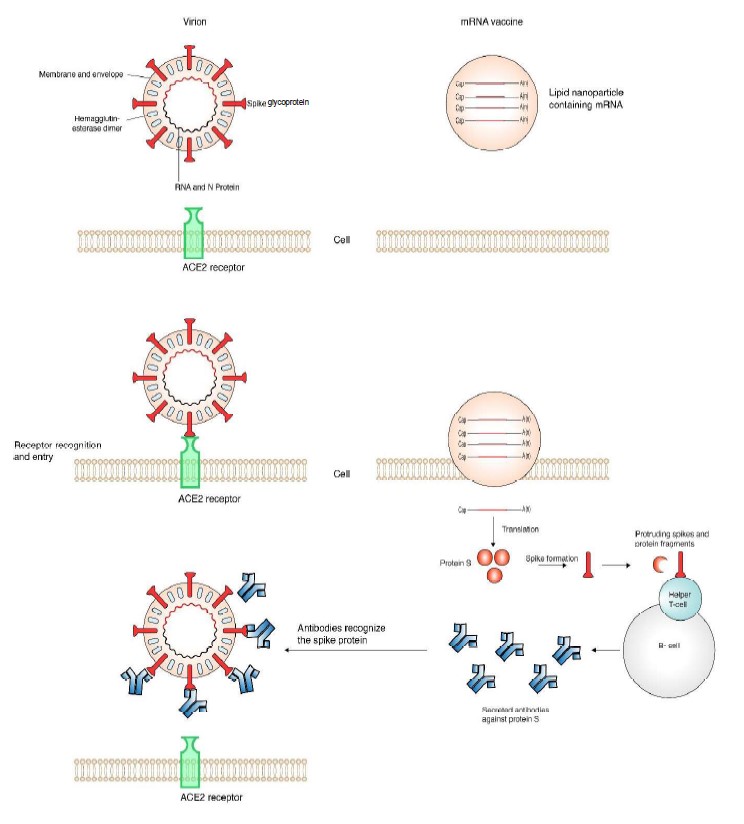
In betacoronaviruses the genome consists of a single RNA, linear, of positive polarity and is approximately 30,000 nt long. The virion is spherical, enveloped, and is 120 nm in diameter (Brian and Baric, 2005; Cui et al., 2019). The genomic RNA is protected by nucleoprotein N in a nucleocapsid. The envelope is formed by the membrane (M) protein and the small membrane protein E. A distinctive feature of the coronavirus virion is the presence of spikes formed by the glycoprotein S (protein S) ( Figure 2) (Lan et al., 2020; Walls et al., 2020; Wrapp et al., 2020a).
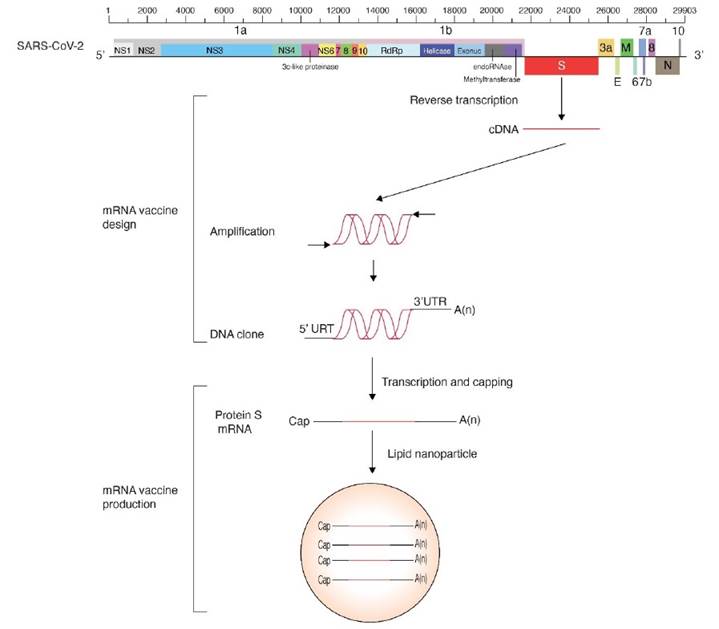
The coronavirus genomic RNA (Figure 3) is capped, polyadenylated and encodes multiple cistrons in open reading frames 1 (ORF1a) and 1b (ORF1b) joined by a ribosomal frameshift. Polyproteins 1a and 1ab are processed by papain-like proteinase NSP3 and 3C-like proteinase NSP5 to form the viral RNA-dependent RNA polymerase and several non-structural proteins necessary for RNA replication. M, E, S and other structural proteins are expressed from subgenomic RNAs co-terminal with the 3’ end, and contain a 5’ leader that is 65 to 89 nt long (Brian and Baric, 2005).
Entry into the cell
Entry into the cell is mediated by protein S spikes on the virion surface that interact with cellular receptors. The process is facilitated by entry cofactors (Gallagher and Buchmeier, 2001; Li, 2013; Cantuti-Castelvetri et al., 2020; Yan et al., 2020). Protein S is divided into S1 and S2 subunits cleaved by cellular proteases and cofactors (Millet and Whittaker, 2015; Cantuti-Castelvetri et al., 2020; Coutard et al., 2020; Xia et al., 2020). The receptor binding domain is located at the tip of the S1 head, and mediates recognition and binding to the surface of the receptor ACE2 (Cai et al., 2020). Interactions between protein S and de cellular receptor are critical for cell entry, and highly specific (Cai et al., 2020). Thus, protein S is a determinant of coronavirus host range (Gallagher and Buchmeier, 2001; Zhai et al., 2020).
The mRNA vaccine
For its critical role in cell entry, the spike S protein is the common target for neutralizing antibodies and vaccines (Brochot et al., 2020; Cai et al., 2020). In people infected with coronavirus, neutralizing antibodies are formed against the entire protein S. However, non-neutralizing antibodies are also developed against the S2 subunit (Brochot et al., 2020; Cai et al., 2020).
Vaccines trigger the formation of neutralizing antibodies against protein S, in the absence of infection. Two mRNA vaccines based on protein S have been authorized. Their design is similar and are based on the genome organization and gene expression of coronaviruses (Figure 3). The cistron coding for protein S was cloned using the sequence from the reference isolate Wuhan-Hu-1 (NC_045512.2). A 5’ UTR, a 3’ UTR, and a poly A tail were added to provide stability and enhance translation efficiency. To account for variation in protein S (Becerra-Flores and Cardozo, 2020) prevalent mutations were introduced, and to further enhance translation efficiency, nucleoside modification were introduced (Pardi et al., 2018). The basic design was made by integrating all basic information previously accumulated from SARS-CoV and MERS (Corbett et al., 2020). For delivery purposes, and to avoid degradation, the mRNA is enclosed in a lipid nanodrop that releases the mRNA into the cell. Ribosomes translate the mRNA into protein S that triggers the formation of neutralizing antibodies (Pardi et al., 2018).
Genome variation
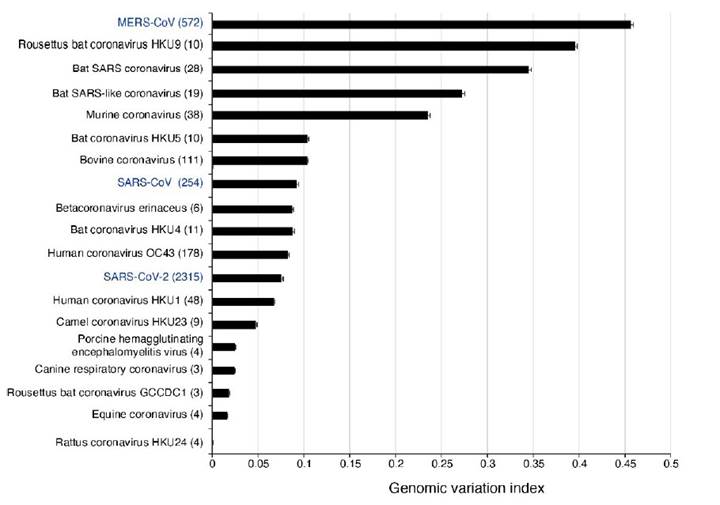
In SARS-CoV-2, mutations have been detected and are being tracked using on-line tools (Hadfield et al., 2018; Fernandes et al., 2020). We recently profiled the genomic variation in all species in the genus Betacoronavirus (LaTourrette et al., 2021). Results showed betacoronaviruses are hyper variable (Figure 4). The most diversity was observed in Rousettus bat coronavirus HKU9, other species infecting bats, and MERS-CoV. In these species, more than 25% of the nucleotides in the genome are polymorphic (Figure 4). The genome of betacoronaviruses consists of 11 to 14 cistrons. The most variable cistron codes for the spike protein S. The lowest variation was detected the cistrons that code for proteins that mediate virus replication: RNA-dependent RNA polymerase, RNA helicase, exonuclease, endo RNAse and methyltransferase, and that are located in open reading frame 1b (LaTourrette et al., 2021).
Protein S variation
Mutations in the genome of SARS-CoV-2 have the potential to affect the precision of diagnostic tests and the efficacy of vaccines. In a recent genome-wide analysis, we showed that hyper variation in protein S is a general feature of betacoronaviruses (LaTourrette et al., 2021). Hyper variation in protein S is evident in betacoronavirus highly pathogenic to humans: MERS-CoV (Figure 5A), SARS-CoV (Figure 5B), and SARS-CoV-2 (Figure 5C). The pattern is also clear in species that infect bats (Figure 5B). Specifically, in SARS-CoV-2 several regions in protein S are hyper variable, including the ACE2 receptor binding domain and the fusion peptide proximal region (Figure 5D).
Betacoronaviruses mainly infect bats and humans (Figure 1). Given the large genetic diversity of bats, and possibly humans, the cellular receptors, proteases, and entry cofactors are likely diverse (Kuo et al., 2000; Cantuti-Castelvetri et al., 2020). Accordingly, protein S hyper variation may provide an evolutionary advantage. Mechanisms driving diversifying selection in protein S may include diversity in cellular receptors, cellular proteases that process the S1/S2 cleavage site, entry cellular cofactors, and antibodies.
Several domains in protein S are intrinsically disordered (LaTourrette et al., 2021): the receptor binding domain and the C-terminal domain 2 in S1, and the fusion peptide proximal region in S2 (Figure 5D). This observation is important because intrinsically disordered proteins mediate functional diversity and interactions with genetically diverse partners such as cellular receptors and entry cofactor in bats and humans (Hebrard et al., 2009; Rantalainen et al., 2011; Charon et al., 2018). Selection for hyper variation and disorder in protein S are consistent with the bat origin of SARS-CoV and SARS-CoV-2 followed by adaptation to humans (Cui et al., 2019; Lu et al., 2020).
In betacoronaviruses, protein S is hyper variable, disordered, mutationally robust (LaTourrette et al., 2021), and is a determinant of host adaptation and host range (Kuo et al., 2000; Muth et al., 2018; Zhai et al., 2020). The emerging model is that in protein S hyper variation provides an evolutionary advantage and is an intrinsic property of betacoronaviruses (LaTourrette et al., 2021).
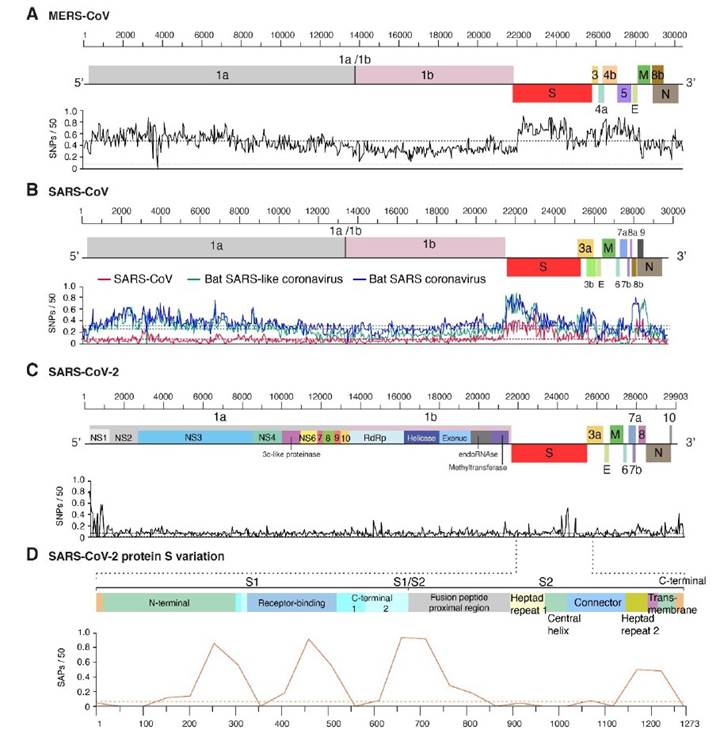
Figure 5 Distribution of genomic variation in human betacoronaviruses. Single nucleotide polymorphisms were plotted with respect to the genome. A 99% confidence interval is indicated as a horizontal line. A) MERS-CoV. B) SARS-CoV. Two species infecting bats are included for comparison. C) SARS-CoV-2. D) Single amino acid polymorphism in SARS-CoV-2 protein S. Domains are annotated and color coded. Modified from LaTourrette et al. (2021).
Antibodies againts protein S
In infected cells, neutralizing antibodies are developed against protein S (Brochot et al., 2020; Cai et al., 2020). The receptor-binding domain is a critical antigen (Noy-Porat et al., 2020). Additionally, non-neutralizing antibodies against protein S fragments of subunit two are also present (Brochot et al., 2020; Cai et al., 2020). Non-neutralizing antibodies may serve as decoys the reduce biogenesis and targeting efficiency of neutralizing antibodies (Cai et al., 2020). Thus, the hyper variation of protein S might be a mechanism for betacoronaviruses to escape the immune system.
Variation in protein S and implications for vaccine use
Vaccines against SARS-CoV-2 induce neutralizing antibodies against protein S (Figure 2) (Cai et al., 2020; Wrapp et al.; Yuan et al., 2020). Hyper variation in protein S has potential to reduce efficacy of vaccines by multiple mechanisms. In an infected individual, new virus variants might be generated and have been detected (Jary et al., 2020) with potential to escape neutralizing antibodies. Furthermore, antibodies developed after vaccination are a selection agents with potential to favor virus variants that can escape neutralizing antibodies (Baum et al., 2020).
Under this scenario, if SARS-CoV-2 remains genetically stable, vaccines will be efficient (Dearlove et al., 2020), antibody-based diagnostic test highly reliable, and infected people who develop antibodies will likely acquire immunity to SARS-CoV-2. However, if SARS-CoV-2 differentiates into strains, vaccines will be efficient only against closely related strains, ineffective against diverse strains, and people might be repeatedly infected by SARS-CoV-2.
Re-infection in humans has been confirmed (Tillett et al., 2020), and hyper variation in protein S is a general feature of betacoronaviruses (LaTourrette et al., 2021). These observations predict that adjustments to vaccine design and antibody-based diagnostic tests will be needed. Vaccines administered to people may consist of a cocktail of protein S variants (Baum et al., 2020; Cai et al., 2020). Alternatively, or in addition, the vaccines may need to be re-designed based on SARS-CoV-2 population dynamics, structure and their geographic distribution (Korber et al., 2020; Taboada et al., 2020; LaTourrette et al., 2021).
It is likely that SARS-CoV-2 will accumulate mutations for efficient replication and differentiate into biological strains as the virus faces selection pressure from genetically different human populations (LaTourrette et al., 2021). Multiple lines of evidence support this model. Despite not reaching pandemic levels, protein S accumulated large numbers of mutations in MERS-CoV and SARS-CoV (Figure 5). SARS-CoV-2 variants infecting the same individual have been detected (Tillett et al., 2020; Jary et al., 2020), and recurrent mutations in open reading frame 1ab, and cistrons coding for NSP6 and protein S have been identified (van Dorp et al., 2020). Additionally, contrasting mutations have also be described. In Mexico, SARS-CoV-2 population were grouped into clades (Taboada et al., 2020), and in Arizona, a 27-amino acid deletion was detected in protein 7 (Holland et al., 2020).
The first genome of the SARS-CoV-2 came from the strain initially described in China, (Wuhan-Hu-1 NC_045512.2). The cistron coding for protein S contains residues that are compatible, but not optimal, for binding human receptor ACE2 (Wan et al., 2020). Thus, there is potential for protein S to accumulate mutations for more efficient entry into human cells. Consistent with this model, a D614G mutation makes the virus more transmissible, more pathogenic to humans (Becerra-Flores and Cardozo, 2020), and has replaced the initial strain (Long et al., 2020; Volz et al., 2020). The D614G mutation and others in the receptor binding domain reduce affinity to monoclonal antibody CR3022 (Long et al., 2020). This is consistent with a role for protein S variation in escaping from neutralizing antibodies.
Future challenges
Multiple lines of evidence support the model that SARS-CoV-2 is mutating (Forster et al., 2020; Korber et al., 2020; Phan, 2020a), and that as a group betacoronaviruses are hypervariable and variation mainly accumulates in protein S (LaTourrette et al., 2021). This variation has the potential to affect the both the efficacy of vaccines and the reliability of antibody-based diagnostic test. Collectively this information predicts that vaccine design and deployment will be based on fundamental understanding and characterization of proteins S, and other genes, in the SARS-CoV-2 genome in a combination of factors such as human population genetics, age groups, health underlying conditions, geographical and regional boundaries. Characterizing the genetic structure of SARS-CoV-2 at fine scale, and translating this variation into the design and deployment of SARS-CoV-2 vaccines is one of the main challenges. To answer this challenge, it will be essential profile the genetic structure of the virus in different parts of the world, in human populations of different genetic backgrounds, and before and after administration of the SARS-CoV-2 vaccines

