











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
Congenital heart defects (CHDs) are anatomic abnormalities of the heart and great vessels that arise during prenatal development1-3. CHDs as a group corresponds to the most common congenital defect, they are a frequent cause of consultation and represent a public health issue1-4. The world incidence of CHDs varies from 6 to 11.1:1000 live newborns (LNB)5; it has been considered that about 1% of LNB suffer from some form of CHD, and this percentage could reach 3% if minor heart anomalies such as bicuspid aortic valve are taken into consideration. CHDs can be diagnosed during prenatal consultation and it has been reported a high in utero mortality depending on the type of alteration present6-8. CHDs frequency also may vary regarding the gender as, for example, atrial septal defects (ASD) are more frequently reported in females; meanwhile, conotruncal heart defects (CTHD) are more common in males3.
Every year an estimated of 20,000 children are born in our country with a CHD2, during 2001 they corresponded to the second-ranked among the death causes in the population under 5 years of age, the fifth place in the 5-9 years of age group, the sixth in the 10-14 years of age, and the 10th cause of disease in the 15-19 years of age group9. In 2013, 24% of childhood mortality in Mexico was due to congenital diseases, with CHDs corresponding to 55% of death causes in children under one year of age10. These data underline the importance of establishing an adequate CHD diagnostic screening, their register, and follow-up to avoid sub-registration and to consider medical attention requirements, among other goals.
CHDs clinical approach is a complex issue with several facets, from establishing a diagnosis and management, identifying etiological factors, to offer genetic assessment, these issues require the expertise of a multidisciplinary medical team. In this sense, genetics is the science that studies the patterns of inheritance, the mechanisms that regulate them, and their consequences in the phenotype. Medical genetics is the application of genetics in medicine and includes to identify the etiology of inherited diseases and their management, the biological variability of the human being, and how these factors are related to the health and disease processes, among other aspects11. Genomics is one of the areas of genetics which carries out, from a structural and functional perspective, the study of the genomes. Genomic technologies are applied to analyze the function, as a whole, of all the genes in the genome and their interactions regarding a given phenotype12. The study of CHDs from the medical genetics perspective includes the study of the effects that the expression of several different genes may have in the normal and pathological heart development. The analysis of these aspects with the technologies available nowadays offers an opportunity to determine the etiology and the possible phenotype-genotype associations, weighting in the effect that the environment may have; it also gives us the possibility to design research for new diagnostic approaches and methods, treatments, and to offer genetic assessment to the patients and their families.
Frequency of the congenital heart diseases
CHDs may occur as an isolated disease, associated with other malformations and/or physiological alterations, or as part of a known clinical syndrome. The frequency of isolated CHDs varies depending on the type of the specific malformation, for instance, it is considered that the most common CHDs are the ventricular septal defects (VSD), and 80% of them correspond to the perimembranous VSD type1. CTHDs represent 15-20% of the CHDs, their clinical manifestations include cyanosis and hypoxia, there is also disorganization of the structure of the ventricles, the aorta, and the pulmonary artery. CTHDs include the tetralogy of Fallot (TOF), double-outlet right ventricle, transposition of the great arteries (TGA), interruption of the aortic arch (IAA), and pulmonary atresia (PA), among other anomalies13.
It has been observed that the frequency of the CHDs in our country changes accordingly to the region or state reviewed. Mendieta-Alcántara et al.14 found that the most frequent CHD in two hospitals in Mexico's State in central Mexico was persistent conduct arteriosus (PCA), and in the specific group of pediatric patients born at term, the most frequent CHDs were interauricular and interventricular communication followed by PCA. Castillo-Espínola et al.2 analyzed the CHDs in Yucatan State in the Southwest of Mexico and reported that the most frequent alteration identified in that region was also PCA (37.6%), followed by TGA. In a study carried out in our institution during a 21-years period15, CHDs were identified in 125 autopsy procedures of pediatric patients; in 55% of the reports the CHDs corresponded to isolated defects, and 23% of them were part of a genetic syndrome, the rest was classified as idiopathic. It is important to consider that when other malformations are identified in organs different from the heart, in up to 30% of the patients, a CHD may also be present. Therefore, in patients with dysmorphic characteristics and/or growth and development alterations, it is important to discard the presence of a CHD. If it is confirmed, its type and clinical characteristics could, in turn, lead the way for a genetic syndrome diagnosis1.
Etiology of the CHDs
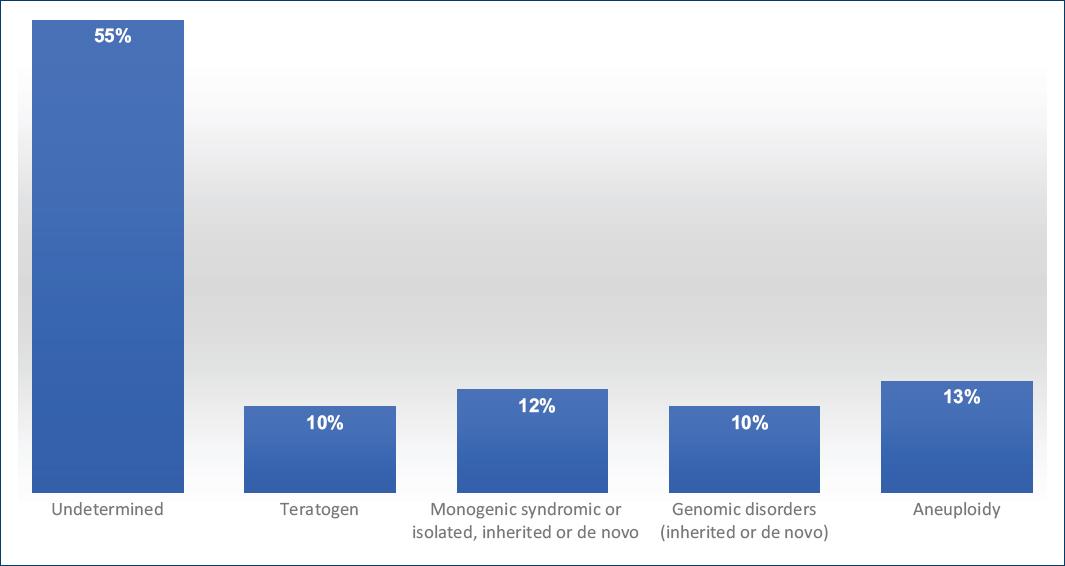
The etiology of most isolated CHDs is considered multifactorial with the participation of genetic and environmental factors. CHDs could be due to the additive effect of both, or to the major effect of one of them in the phenotype, such as the susceptibility given by a pathogenic genetic variant, or due to the exposition to a teratogenic agent. The CHDs that form part of a syndrome may be due to chromosomopathies, monogenic syndromes (MS), and/or to structural rearrangements of the genome. The percentages of the different etiologies implicated in the CHDs are shown in figure 16,16.
CHDs and chromosomal aberrations
CHDs can be associated with numerical or structural chromosomal anomalies, among them the aneuploidies, which are defined as an alteration in the chromosome number that is not an exact multiple of the haploid chromosome number, that is 23 in humans. The aneuploidies can affect the autosomes, as is the case of the trisomy 21 (T21) or Down syndrome; or to the sexual chromosomes, as is the case of Turner syndrome with an X chromosome monosomy. The incidence of aneuploidies in LNB with CHDs is 9-18%16.
The T21 syndrome in 95% of the cases is due to a regular trisomy, in 4% is caused by a Robertsonian translocation or by a 21q isochromosome, in 1-2% is a mosaic, and in a very low proportion is due to the duplication of the chromosomal 21 critical region. The frequency of T21 syndrome in our country is 1/650 LNB17. The CHDs prevalence in T21 syndrome patients is 45-50%; the most frequently identified types are the atrioventricular defect, persistent patent ductus arteriosus (PDA), ASD, and VSD. It has been reported that these frequencies may vary depending of the region considered as, for example, in Latin-American the CHD most frequently reported is the ASD (24%), in Europe is the atrioventricular septal defect (40-80%); and in a Mexican patient's cohort PDA was the most frequently reported (22%)18.
Turner syndrome has a frequency of 1:2500 female newborn, in 50% of the cases, it is caused by an X chromosome monosomy with a 45,X karyotype. The phenotype in this syndrome can vary depending on the age of the patient and includes congenital lymphedema in hands and feet, short stature, pterygium colli, cubitus valgus, and gonadal dysgenesis, among others. Between 35% and 40% of Turner syndrome patients may occur a cardiovascular malformation. The most common alteration is the bicuspid aortic valve in 16% of the cases, and coarctation of the aorta (CoA) is identified in 11-14%. In adult Turner syndrome, patients may occur complications, such as aortic dissection (1-2%), systemic hypertension (30-50%), and ischemic heart failure19.
In the study carried out by Hartman et al., in a group of LNB with CHD, the most common chromosomopathies identified were T21 syndrome in 52.8% of the cases, Trisomy 18 or Edwards syndrome in 12.8%, and Trisomy 13 or Patau syndrome in 5.7%; the 22q11.2 microdeletion syndrome was present in 12.2%; it was concluded that 1 in 8 patients with CHD had some type of chromosomopathy20. The CHDs may be part of the malformations diagnosed in different syndromes resulting from non-balanced structural chromosomal aberrations which produce partial monosomies or trisomies. Microdeletions or microduplications associated to copy number variants (CNV) of DNA sequences can cause disorders such as the mentioned deletion 22q11.2 syndrome (MIM 192430) which is particularly associated to CTHD (Table 1)16,21,22.
Table 1 Chromosomal syndromes and genomic disorders with congenital heart disease
| Syndrome | Gene(s)* | MIM | % CHD | Type of CHD | Main clinical characteristics |
|---|---|---|---|---|---|
| Aneuploidy | |||||
| Trisomy13 Patau syndrome | - | - | 80 | CTHD, TOF, VSD, ASD, DORV, CoA | Aplasia cutis, holoprosencephaly, microphthalmia, CLP, polydactyly, ID |
| Trisomy 18 Edwards syndrome | - | - | 80-100 | VSD, ASD, TOF, AVSD, DORV, PDA | Growth and developmental restriction, clenched hands with overlapping fingers, club foot, DI |
| Trisomy 21 Down syndrome | DYRK, COL6A1, COL6A2, ** modify by CRELD1, FBLN2, FRZB, GATA5 | 190685 | 45-50 | AVSD, VSD, ASD, TOF | Characteristic facies, hypothyroidism, hypotonia, ID, among others |
| X Monosomy Turner syndrome | **modifiers SLC2A2, SLC2A14 | - | 35-40 | BAV, AS, CoA, HLHS, SD | Pterygium colli, cubitus valgus, short stature, gonadal dysgenesis |
| Genomic Disorders | |||||
| del 1p36 | PRDM16 | 607872 605557 | 70 | SD, VSD, ASD, AVSD, TOF, CoA, PDA, CM, Ebstein anomaly. | Craniofacial and brain alterations, sensorineural deafness, seizures, ID |
| del 1q21.1 | GJA5 | 612474 121013 | 10-25 | CoA, BAV, SVAS, SD, CTHD | Microcephaly, developmental delay, ID, autism, and schizophrenia |
| dup 1q21 | GJA5 | 612475 121013 | 20 | TOF | Macrocephaly, developmental delay |
| del 4p16.3 Wolf Hirschhorn syndrome | NSD2/WHSC1 | 194190 602952 | 50 | ASD, PS, VSD, PDA | Prenatal and postnatal growth restriction, psychomotor developmental delay, ID, characteristic facies and seizures |
| del 5p15.3 Cri du chat syndrome | - | 123450 | 29 | PDA, SVD, ASD, TOF, PA | Microcephaly, round facies, hypertelorism, micrognatia, typical high-pitched cry, ID |
| del 7q11.23 Williams syndrome | ELN | 194050 130160 | 75-82 | SVAS, PPS | Characteristic facies, hypercalcemia, social personality, ID |
| dup 7q11.23 | ELN | 609757 130160 | 20 | PDA | Language alterations, cognitive defects from normal, to ID to autism to, craniofacial anomalies |
| del 8p23 | GATA4 | - 600576 | 75-94 | AVSD, ASD, TOF, Ebstein anomaly | Congenital diaphragmatic hernia, Microcephaly, developmental delay |
| del 11q23 Jacobsen syndrome | ETS1 | 147791 164720 | 56 | SD, DORV, HLHS, CoA | Thrombocytopenia, dysmorphic features, anal atresia or stenosis, intestinal malrotation, ID |
| del 16p11.2 | - | 611913 | - | BAV, AS | Macrocephaly, CNS and vertebrae anomalies, ID, autism, seizures |
| del 17p11.2 Smith-Magenis syndrome | RAI1 | 182290 607642 | 30 | PA, TOF, SD, APVR, PS | Dysmorphic characteristics, ID, self-injurious behavior |
| dup 17p11.2 Potocki-Lupski syndrome | RAI1 | 610883 607642 | 40 | SD, heart conduction abnormalities, HLHS, BAV | ID, autism, hypotonia, congenital malformations |
| del 22q11.2 Velocardiofacial or DiGeorge syndrome | TBX1 *Modifier SLC2A3 | 188400 192430 | 7580 | CTHD, PTA, IAA-B, TOF, VSD, DORV, IAAA | Variable clinical spectrum, characteristic facies, thymus hypoplasia, speech defects immune and endocrine abnormalities, thrombocytopenia, and characteristic facial features, SMCP, cognitive skills, and psychiatric alterations |
| del 22q11.2 distal | CRKL, MAPK1 | 611867 | 50 | VSD, PTA, BAV, hypoplastic aortic arch, PS | Developmental delay, facial dysmorphism |
| dup 22q11.2 | TBX1 | 608363 | A few cases described | TGA, DORV, CoA | ID, facial dysmorphism |
| 22q partial tetrasomy Inv dup (22)(q11) Cat eye syndrome | 115470 | 50-67 | APVR, TOF, PS, TA, HLHS | Coloboma, anorectal malformations, and preauricular tags, renal malformations, biliary atresia, and malrotation of the gut | |
*genes with loci in the implicated chromosomal regions and are associated with the development of the CHDs in these entities.
**Genes with loci in other regions of the genome and which variants modify the phenotype or help to the presentation of the CHDs
APVR: anomalous pulmonary venous return; AS: aortic stenosis; ASD: atrial septal defect; AVSD: atrioventricular septal defect; BAV: bicuspid aortic valve; CLP: cleft lip and palate; CM: cardiomyopathy; CNS: central nervous system; CoA: coarctation of the aorta; CTHD: conotruncal heart defects; DORV: double outlet right ventricle; HLHS: hypoplastic left heart syndrome; IAAA: isolated abnormalities of the aortic arch; IAA-B: interrupted aortic arch type B; ID: intellectual disability; PA: pulmonary atresia; PDA: persistent ductus arteriosus; PPS: peripheral pulmonary stenosis; PS: pulmonary stenosis; PTA: persistent truncus arteriosus; TA: truncus arteriosus; TGA: transposition of the great arteries; TOF: tetralogy of Fallot; SD: septal defects; SMCP: submucous cleft palate; SVAS: supravalvular aortic stenosis; VSD: ventricular septal defect.
CHDs and genomic disorders due to CNVs
Chromosomal alterations that involve CNVs include the gain or the loss of a segment of DNA which originates microdeletions or microduplications of variable length. Most of them are submicroscopic and cannot be detected with a GTG banding karyotype. These anomalies affect any part of the genome and can be recurrent or not; frequently, the implicated segments are flanked or limited by low copy repetitive sequences. The diseases derived from these chromosomal alterations are known as genomic disorders, as the architecture per se of the genome is the factor that determines its presentation by favoring the occurrence of microdeletions and/or microduplications by events of non-allelic homologous recombination; these regions, in general, include several dose sensitive genes, a situation that modifies the associated phenotype23.
The most frequent genomic disorder in humans is the microdeletion 22q11.2 (del22q11.2) syndrome, also known as velocardiofacial syndrome (MIM 188400, 192430), which affects to 1:2000-1:4000 LNB. The del22q11.2 syndrome shows a wide phenotypic spectrum; the main clinical data are the characteristic facies, submucous cleft palate, and CTHD. The clinical manifestations may not be present in all the patients with this diagnosis; on the other hand, some of them may correspond to its most severe form of presentation and correspond to the DiGeorge syndrome that forms part of the spectrum of the del22q11.2 syndrome and includes thymus hypoplasia or agenesia. The chromosomal deletion in 22q11.2 is 3 megabase in length in most patients and includes almost 40 genes, among them TBX1 (MIM 602054), which has been associated with the presentation of CHD. The del22q11.2 syndrome by itself is associated with 0.5-1.9% of all the CHDs13, and almost 75-80% of the patients with del22q11.2 syndrome present a CHD6,13,24; in a Mexican pediatric patient's cohort, this percentage was even higher (97%)25. The main CHDs described in del22q11.2 syndrome are CTHD with TOF (20% of the cases), IAA type B (13%), persistent truncus arteriosus (6%), and VSD19.
Another syndrome associated with CNVs that include CHDs among its clinical characteristics are the Williams-Beuren syndrome (WBS, MIM 194050); it has a frequency of 1:10,000 LNB and is caused by an interstitial microdeletion in 7q11.23. This syndrome has a characteristic facies, peculiar behavior, hypercalcemia (15%), and CHD in 80% of the cases, being supravalvular aortic stenosis the cardiopathy most frequently diagnosed. One of the genes that are affected by the microdeletion is ELN (MIM 130160) which encodes elastin, the haploinsufficiency of this protein is associated to the cardiovascular pathology observed in the WBS; about 50% of these patients will develop hypertension as they grow up6,19.
These two syndromes, del22q11.2 and WBS, which are caused by a microdeletion, have as their counterparts the syndromes produced by the microduplication of the same chromosomal segments, which can also present CHDs. Many other genomic disorders have been described, and an important percentage of them include CHDs (Table 1)16,22. Pathogenic CNVs have been identified in 15-20% of the patients with syndromic CHDs, and in 4-14 % of patients with isolated CC21. In general, terms, when there is a clinically suspected genomic disorder, the confirmation of the diagnosis is done by a molecular karyotype based in a microarray analysis to be able to identify the genetic etiology in accordance with the associated phenotype observed in the patient.
CHDs and MS
CHD can be one of the clinical characteristics of the phenotype of genetic diseases originated by monogenic alterations with a Mendelian inheritance pattern. The presence of a given CHD in these disorders is relevant as they can support diagnosis and can also influence the clinical management and follow-up of the patient. The genes implicated in these syndromes encode proteins with diverse functions, among them are transcription factors implicated in the development of the heart or other organs, as happens with TBX5 in the Holt-Oram syndrome; other genes that cause MS with CHDs codify for proteins that participate in signal transduction pathways, as pathogenic variants in JAG1 or NOTCH2 do in the Alagille syndrome (Table 2)6,21-25.
Table 2 Monogenic syndromes with congenital heart defects
| Syndrome | Genes | MIM | Pattern of inheritance | % CHD | Type of CHD | Associated clinical characteristics |
|---|---|---|---|---|---|---|
| Transcription factors | ||||||
| Holt Oram | TBX5 | 142900 601620 | AD | ASD, VSD, MVA, HLHS, TAC, CoA, AS, PDA | Upper extremities anomalies | |
| Signal transduction | ||||||
| Alagille | JAG1, NOTCH2 | 118450 601920 600275 | AD | > 90 | PS, VSD, ASD, AS, CoA TOF | Cholestasis, butterfly vertebra, ocular alterations, characteristic facies |
| AdamsOliver | NOTCH1 DLL4 | 616028 190192 616589 605185 | AD | 20 | BAV, CoA, HLHS, VSD, TOF, TA | Aplasia cutis, transverse terminal limb reduction defects, syndactyly, brachydactyly, hemangioma |
| RASopathies | 60-90 | |||||
| Noonan | PTPN11, SOS1, KRAS, RAF1, BRAF, SHOC2, NRAS, CBL, SOS2, RIT1, y LZTR1 | 163550 610733 609942 611553 613706 602775 613224 165360 616554 615355 616564 | AD | 80 | PS ASD, VSD, TOF, CoA, HCM | Dysmorphic facies, short stature, broad or webbed neck, chest deformity, variable learning disability |
| Leopard Multiple lentigines syndrome | PTPN11 RAF1 BRAF | 151100 611554 613707 | AD | 80-90 | HCM, PS, heart conduction abnormalities | Lentigines, hypertelorism, genital abnormalities, retardation of growth, sensorineural deafness |
| Cardiofaciocutaneous | BRAF KRAS MAP2K1 MAP2K2 | 115150 190070 176872 601263 | AD | 75-80 | PS, ASD, HCM | ID, characteristic facies, ectodermal abnormalities |
| Costello | HRAS | 218040 | AD | 44 | PS, heart hypertrophy, heart arrhythmia, TA | Coarse facies, short stature, feeding difficulty, ID |
| Syndrome | Genes | MIM | Pattern of inheritance | % CHD | Type of CHD | Associated clinical characteristics |
| Ciliopathies | ||||||
| Primary ciliary dyskinesia 1 Kartagener | DNAI1 | 244400 604366 | AR | 50 | HTX, SI, dextrocardia | Male infertility, bronchiectasis, SI |
| Bardet Biedl 2 | BBS2 | 615981 | AR | 30-50 | ASD, BAV, DCM | ID, Polydactyly, retinitis pigmentosa, kidney anomalies, obesity |
| McKusick-Kaufman | MKKS | 236700 604896 | AR | A few cases described | ASD, VSD, CoA, PDA | Hydrometrocolpos, postaxial polydactyly |
| Cromatinopatías | ||||||
| CHARGE | CHD7 SEMA3E | 214800 | AD | 85 | TOF, DORV, AAA, CTHD | Dysmorphic facies, coloboma, choanal atresia, inner ear, genital, and retinal abnormalities |
| Cornelia de Lange | NIPBL SMC1B SMC3 RAD21 HDAC8 | 122470 300590 610759 614701 300882 | AD XLD AD AD XLD | 33-40 | PS, VA, VSD, AVSD, BAV | Dysmorphic facies, ID, limb abnormalities, and hirsutism, synophrys, long eyelashes |
| Kabuki | KMT2D/MLL2 KDM6A | 147920 300867 | AD XLD | 70 | CoA, VSD, ASD | Dysmorphic facies, cleft palate, vertebral abnormalities, seizures, hypotonia |
| Kleefstra (del 9q34.3)* | EHMT1/KMT2C | 610253 617768 607001 606833 | AD | 40 | ASD, VSD, TOF, CoA, BAV, PS | Hypotonia, ID, characteristic facies, synophrys, carp mouth with macroglossia |
| Koolen de Vries (del 17q21.31)* | KANSL1/KAT8 | 610443 612452 | AD | 39 | SD, PS, BAV | ID, hypotonia, characteristic facies, genitourinary anomalies, seizures |
| Roberts SC Phocomelia | ESCO2 | 268300 269000 | AR | 20 | AVSD, PTA, VSD, ASD, PDA | Limb deformities, craniofacial anomalies, growth retardation |
| Rubinstein Taby (del 16p13)* | CREBBP EP300 | 180849 613684 | AD AD | 33 | ASD, VSD, PDA, CoA, HLHS | ID, broad thumbs and great toes, characteristic facies |
| Sotos (del 5q35)* | NSD1 | 117550 606681 | 8 | ASD, VSD, PDA, | Dysmorphic facies, macrocephaly, hypospadias, macrosomia, seizures | |
*These syndromes may be originated by a genomic disorder which includes the haploinsufficiency of the responsible gene or maybe due to a mutation in such gene. AAA: aortic arch anomalies; AD: autosomic dominant; AR: autosomic recessive; AS: aortic stenosis; ASD: atrial septal defect; AVSD: atrioventricular septal defect BAV: bicuspid aortic valve; CTHD: conotruncal heart defects; CoA: coarctation of the aorta; DCM: dilated cardiomyopathy; DORV: double outlet right ventricle; ID: intellectual disability; HCM: hypertrophic cardiomyopathy; HLHS: hypoplastic left heart syndrome; HTX: heterotaxy; MVA: mitral valve defect; PDA: persistent ductus arteriosus; PS: pulmonary stenosis; PTA: persistent truncus arteriosus; SI situs inversus; SD: septal defects; TA: truncus arteriosus; TAC: truncus arteriosus communis; TOF: tetralogy of Fallot; VA: valvular anomalies; VSD: ventricular septal defect; XLD: X-linked dominant.
The mutations or variants that affect different genes that participate in the same pathway may originate diseases that share some clinical characteristics, including CHDs. This phenomenon is observed in the group of disorders known as RASopathies, which have an autosomic dominant (AD) pattern of inheritance. These diseases are caused by alterations of the Ras/MAPK (mitogen-activated protein kinase) signal transduction pathway. Among the RASopathies are Noonan (MIM 163950), cardiofaciocutaneous (MIM 115150), and Costello syndrome. They share some characteristics, such as a particular facies, growth, and heart alterations (Table 2)26,27.
Another group of monogenic disorders that have CHDs are the ciliopathies, diseases caused by abnormalities in the structure and function of cilia, their clinical characteristics vary accordingly to the specific disease. Cilia have an important participation in the development of the heart to establish the characteristics of each side and of the left-right asymmetry. One of the ciliopathies is the primary ciliary dyskinesia associated with CHD in 30-50% of the patients and includes heterotaxy (Table 2)6,22.
Isolated CHDs and monogenic variants
Epidemiological analyses have suggested that genetic factors are a predominant cause of isolated CHDs; nonetheless, the environmental exposition to diverse teratogenic agents cannot be excluded as participants in their etiology. Taken this situation into account, a multifactorial pattern of inheritance for the isolated CHDs has been proposed; however, trying to explain the expected recurrence risk for this type of CHDs is very complex. In some families, their percentages are higher than the expected recurrence risks predicted accordingly to the polygenic and multifactorial models of inheritance. The study of these families has allowed the identification of pathogenic genetic variants, which have a Mendelian inheritance pattern and show an incomplete penetrance in the pedigree. They represent predisposition effects for CHDs and show phenotypic heterogeneity in familial cases, and also in de novo presentations. This complexity may be due to the added action of non-genetic factors such as the exposition to environmental agents, to epigenetic alterations or to stochastic factors (meaning random in nature) effects28,29; but the effect of modifying genes with a relevant roll in the development of isolated and syndromic CHDs cannot be discarded8.
It has been estimated that up to 2% of the isolated CHDs cases are related to a punctual mutation in a given gene, for example, NKX2.5 has been associated with the presentation of ASD, CTHD, hypoplastic left heart syndrome (HLHS), and cardiac conduction defects21,22. Some of the genes with genetic variants that have been associated with CHD with an isolated, familial, or sporadic presentation are listed in table 36,21-25.
Table 3 Genes with variants in sequence or in CNVs that have been associated with the development of isolated congenital heart disease in de novo or/and familial cases
| Gen | MIM | Locus | Type of CHD |
|---|---|---|---|
| Transcription factors | |||
| GATA4 | 600576 | 8p23.1 | ASD, VSD, AVSD, TOF |
| GATA5 | 611496 | 20q13.33 | AVSD |
| GATA6 | 601656 | 18q11.2 | ASD, AVSD, TAC, TOF |
| NKX2.5 | 600584 | 5q35,1 | ASD, VSD, TAC, TOF, HLHS |
| NKX2.6 | 611770 | 8p21.2 | CTHD, TAC |
| PRDM16 | 605557 | 1p36.32 | DCM, NCLV |
| TBX1 | 602054 | 22q11.21 | TOF |
| TBX5 | 601620 | 12q24.21 | ASD, VSD |
| TBX20 | 606061 | 7p14.2 | ASD |
| ZFPM2 | 603693 | 8q23.1 | CTHD, TOF |
| Transduction signal | |||
| NODAL | 601265 | 10q22.1 | Heterotaxy |
| GDF1 | 602880 | 19p13.11 | TGA, TOF |
| NOTCH1 | 190198 | 9q34.3 | Aortic valve anomalies |
| JAG1 | 601920 | 20p12.2 | TOF |
| Other | |||
| ELN | 130160 | 7q11.23 | SVAS |
| GJA1 | 121014 | 6q22.31 | AVSD, HLHS |
| MED13L | 608771 | 12q24.21 | TGA |
ASD: atrial septal defect; AVSD: atrioventricular septal defect; CTHD: conotruncal heart defects; DCM: dilated cardiomyopathy; HLHS: hypoplastic left heart syndrome; TAC: truncus arteriosus communis; TGA: transposition of the great arteries; TOF: tetralogy of Fallot; NCLV: non-compacted left ventricle; SVAS: supravalvular aortic stenosis; VSD: ventricular septal defect.
CHDs associated with the exposition to teratogenic agents
As mentioned, the development of the heart is a complex process and requires the coordinated action of multiple genes, which are susceptible to the effect of the environment, which may have a teratogenic factor and originate a CHD. Among this type of environmental element is, for example, the infection by rubella virus in the mother that can cause CHDs in the fetus, among other alterations. Another aspect with relevant participation in the presentation of CHDs is the lifestyle, as smoking, alcoholism, and obesity in the mothers have been recognized as factors that modify the risk of presenting CHD in their descents. Chronic maternal diseases associated with the presentation of CHDs in the fetus are diabetes, hypertension, and epilepsy30. The pregestational maternal diabetes has a risk of developing a CHD in the fetus of 3-5%; the prescription of anticonvulsant drugs for maternal epilepsy, increases the risk of CHD to 1.8%; and the exposition to other drugs, such as the retinoic acid, may increase the risk to levels as high as 8-20%7.
Another chronic maternal disease that increments the risk of a CHD presentation in the fetus is the phenylketonuria, if there are elevated maternal serum levels of phenylalanine (> 15 mg/dL) as they are associated with a 10 to 15-fold increased risk of CHD. It is important to underline that the pregestational evaluation of both parents taking into consideration the risk factors of each of them (including the environmental and hereditary characteristics) has as its main goal a close follow-up of the pregnancy to allow it to develop in a protected and secure manner, in the best possible conditions, to prevent complications.
CHDs and epigenetics
The term epigenetics refers to all the stable and inherited chemical modifications, which change the chromatin structure and affect the expression of the genes without changing the sequence of the nucleotides of the DNA. The epigenetic changes are given through the post-translational modifications of the histones and by changes in the methylation state of the cytosine residues of the CpG dinucleotides located in specific sites of the genomic DNA. The enzymes that introduce these changes in chromatin are known as "writers." The epigenetic modifications give place to different structures in the chromatin due to interactions with molecules that recognized the modifications and are called "readers," which, in turn, allows or impedes the access of the transcription machinery to the DNA. The epigenetic changes can be reversed by the action of enzymes that act as "erasers"31.
Multiprotein remodeling complexes of the chromatin also participate in the epigenetic changes that modified the chromatin from an active state in transcription to an inactive one or vice versa; also, non-coding RNAs participate in this process as modulators. The epigenetic process regulates the genetic expression depending on the environmental conditions and confers to an organism the capacity to induce alternative phenotypes derived from the same genotype and with the adequate plasticity to be able to adapt to the environmental conditions through the regulation of the patterns of gene expression31,32.
The epigenetic alterations of the chromatin are associated with a heterogeneous group of more than 50 MS named chromatinopathies, in which a genetic pathogenic variant causes a disruption of one or several components of the epigenetic machinery. These diseases are produced by variants in genes of the "writers" as happens in the Kabuki 1, Sotos, Kleefstra y Rubinstein-Taybi, syndromes among others; of the readers as in the Smith-Magenis and Rett syndromes, or of the erasers as in the Kabuki 2, Townes-Brock, Bohring-Opitz syndromes; and could also occur in remodeling complexes of chromatin as in the CHARGE syndrome (Table 2)32.
The alterations in the epigenetic machinery produce a wide spectrum of clinical data due to a pleiotropic effect. Among their most frequent clinical manifestations are the delayed psychomotor development and intellectual disability, this situation may suggest that a normal epigenetic state is important to conserve the neuronal homeostasis; these diseases also have a wide variety of congenital malformations, including CHDs which are present in an important proportion of patients with Kabuki, Kleefstra, CHARGE, Sotos, and Rubinstain Taby syndromes, among others (Table 2)30. The expression of the genes implicated in the development of the heart can also be modified by epigenetic alterations originated by environmental changes32,33.
CHDs as part of syndromes without a defined etiology
An "association" is defined by the presence of a group of malformations that occurs together more frequently that would be expected by chance. An example of these type of defect is VACTERL association, it has a frequency of 1:7 000-1:40 000 LNB, and its name corresponds to the acronym formed by the malformations included: vertebral defects (V), anal atresia (A), cardiac malformations (C), tracheo-esophageal fistula (TE), renal (R), and limb (L) alterations. The diagnosis of VACTERL is defined by the presence of at least three of the mentioned congenital malformations, another aspect to take into consideration for its diagnosis is that there should be no evidence of other similar condition or a chromosomal alteration; therefore, it is a diagnosis by exclusion of other pathologies. In up to 40-80% of the patients with VACTERL, a CHD can be identified, which can be a severe structural malformation or a functional alteration34.
Comorbidities in congenital heart diseases
The life span of patients with CHD has improved in recent times due to medical advances such as an early diagnosis, including the prenatal, the management of the complications, and corrective surgery techniques. In general, terms and depending on the type of CHD, the overall survival of the patients is higher than 90%. However, their clinical management may require high-cost treatments, and, in some of them, may have a poor prognosis. Sometimes the patients with a CHD may be diagnosed later in life; therefore, a specialized medical consultation for these patients is required, among its objectives is to integrate them in an adequate manner to a productive life. The prevalence of CHD in the adult population is 4.09/1000; in some of these populations, the most frequent lesion was CTHD, including TOF16,35. This situation reflects the fact that the patients with CHDs will require an appropriate medical follow-up since the pediatric age that needs to be continued up to the adult age3.
In this order of ideas, another important aspect to consider in the clinical approach of the patients with CHDs is the future morbidities that they may develop. Some studies have underlined the possible risk that some of these patients show for alterations in the neurodevelopment; this situation may be related to complications present at birth or during a surgical procedure, among other causes. Another consideration is that the altered neurodevelopment and the heart disease in these patients may have a common genetic etiology as occurs in diseases such as chromatinopathies and chromosomopathies, or in the case of the respiratory disease that is present in the ciliopathies. It has also been suggested that having a CHD may signal a risk in adult age to present heart failure, arrhythmia, and pulmonary hypertension16.
The consideration of all these aspects is important for decision-making, for example, the medical risk to consider in particular ages of life as would be a pregnancy in a CHD female patient, taken into account both the physiological condition challenge for the heart, and to offer genetic assessment accordingly to the different genetic etiologies of the CHD36. An intervention that may be of benefice for children with CHD is that, depending on the severity of the case, they could participate in early neurodevelopment stimulation programs that will help in their management37.
Diagnostic methods to determine the etiology of the CHDs and genomic medicine
The karyotype with GTG banding technique is the analysis used to detect chromosomal aberrations; in this study, as a general rule, 30 metaphases are analyzed, this number allows the identification of mosaic alterations, in which the cell line in lower proportion would be in about 17-19%. The karyotype with GTG banding identifies structural chromosomes aberrations in the rank of 3-5 Mb. Smaller structural chromosomal alterations can be detected by molecular studies such as fluorescent in situ hybridization that use marked probes specific for a chromosomal region.
A recent method regarding the evaluation of patients to discard the diagnosis of chromosomopathy is the molecular karyotype by microarrays. Microarray analysis has a resolution power at least 5 times higher than the GTG banding karyotype; however, it has some limitations such as that it cannot detect balanced chromosomal aberrations and that some of the results obtained by this method can be variants of uncertain significance (VUS). Another alternative for the detection of changes in CNVs is the analyses by multiplex ligation-dependent probe amplification (MILPA); this study can simultaneously analyze 25 or more genomic regions associated with CHDs and could also detect specific mutations if the probes are included in the essay38. The clinical evaluation of a patient will give a presumptive diagnosis, and based on it, a decision about the best laboratory study option should be taken, also considering the time needed to obtain a result, the type of information that it may provide, and its cost39.
Genomic medicine, also known as personalized or precision medicine, is defined as the use of the genomic analysis information of an individual to increase the quality of the medical attention12,40. Genomic medicine studies include the presymptomatic identification of genetic markers of predisposition to multifactorial diseases; this information can lead to implement preventive intervention strategies, specific selection of the appropriated pharmacotherapy, and/or the personalized design of a given medical treatment based on the genotype of each particular patient.
Genomic medicine applies also to monogenic diseases, chromosomopathies, and genomic disorders; this analysis has been possible as the laboratory techniques have evolved from the analysis of only one gene to review several genes or panels of genes, and even the exoma (2% of the genome) or whole genome can be analyzed. The genomic techniques include next-generation sequencing that could identify multiple sequence variants and structural variants which can be pathogenic, not pathogenic, or VUS. The clinical interpretation of the VUSs represents a challenge, and different complex statistical and bioinformatics analyses have been carried out in different populations to establish a reference for them and determine their possible participation in the phenotype of a patient41. It is not infrequent that the identified pathogenic variants are private to one given family, and therefore a careful interpretation of the findings is required before considering a phenotype-genotype correlation.
To have a proper clinical evaluation, the clinical practitioner must consider that when a patient with a CHD is examined, the type of the heart anomaly, the stage of development in which it was diagnosed, the age of the patient, the family medical history, other dysmorphic features, and/or other extracardiac anomalies or malformations must be taken in consideration in each individual case. The mentioned information will lead to possible etiologies and/or to identify risk factors involved in the presentation of a CHD, which in turn will have consequences for diagnosis, management, treatment, and genetic assessment (Fig. 2)6,16,25. For example, fetus with CHDs diagnosed by prenatal ultrasound studies has a risk 15-30% higher of presenting a chromosomopathy, depending on the technique that was used7. The exome sequencing analysis has revealed that up to 10% of the isolated CHDs are caused by coding de novo variants, and when extracardiac and/or neurodevelopmental alterations are identified, these techniques allow to identify a cause in up to 20% of the patients6.
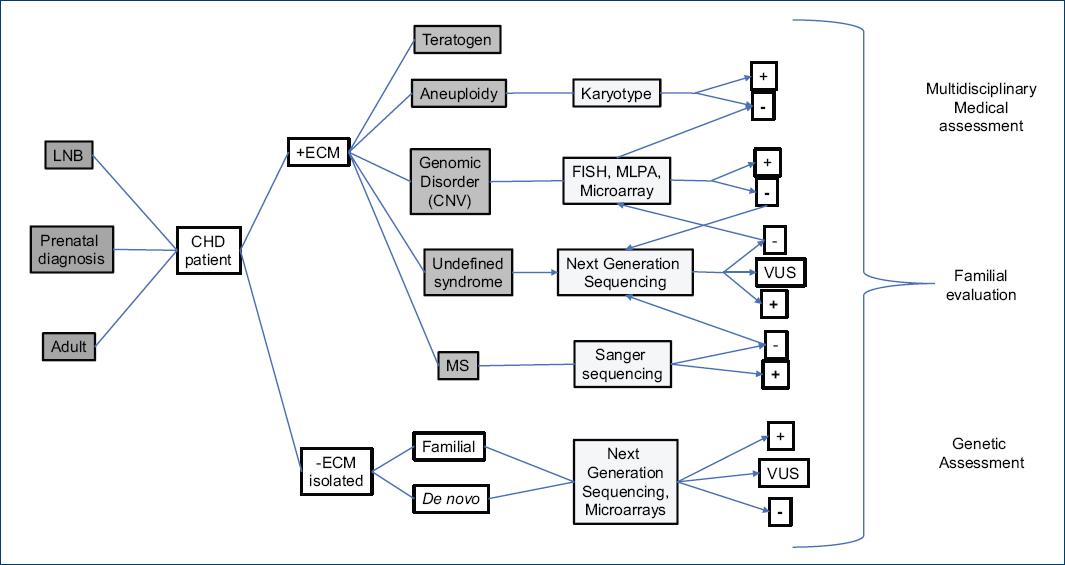
Figure 2 Flow chart for the investigation and genetic diagnosis of the congenital heart defects. +ECM: presence of external congenital malformation(s), −ECM: without external congenital malformation(s), CHD: congenital heart defect, CNV: copy number variant, FISH: fluorescent in situ hybridization, MLPA: multiplex ligation-dependent probe amplification analyses, LNB: live newborn, MS: monogenic syndrome, VUS: variant of uncertain significance.
Recurrence risk in CHDs and genetic assessment
The risk of presentation of an isolated CHD in the general population is < 1%; however, this percentage can be increased by different factors such as hereditary or teratogenic elements22. When evaluating a patient with CHD, among the factors to be considered is if in the family clinical history are other CHD affected members, the type of alteration, and the parentage relation that exists with the propositus. In the case that the mother is who present an isolated heart structural anomaly, the recurrence risk for the descendants is 3-7%, a percentage that in general terms is higher than if the father is who had the alteration, in which case the risk is 2-3%. The recurrence risk for a sibling of a CHD patient with a structural anomaly is 3%, and in particular instances such as HLHS the risk can be increased to 8%5.
The genetic assessment of a syndromic CHD will be considered accordingly to the type of anomaly and the etiological cause that had been established. When considering monogenic disorders such as Alagille, Noonan or cardiofaciocutaneous syndromes, all of them with a CHD among their particular clinical characteristics, these syndromes have an AD inheritance pattern; therefore, the risk of recurrence will be of 50% for the children of a patient with the disease, a risk to be considered in each pregnancy and independently of gender, as both male and females can be affected. When the syndromic CHD in the propositus is classified as a de novo event, the risk of recurrence for the parents of the patient is similar to the risk of the general populations and will be 50% for the descendants of the patient if it is AD; when the pattern of inheritance of the disease is autosomic recessive, as occurs in the ciliopathies, the risk of recurrence for the parents of the patient will be 25%, per pregnancy and independently of the gender of the fetus.
In the case of the genomic disorders such as the del22q11.2, a patient who has to reach a reproductive age can inherit to the descendants either a normal 22 chromosome or the one that has the deletion; therefore, the risk of recurrence will be 50%. In the case of this syndrome, given the wide phenotypic variability and the knowledge that up to 10% of the cases have a family presentation, the study of both parents to confirm or discard a de novo presentation or to determine if one of them have the 22q11.2 deletion or a balanced chromosomal rearrangement that involves this chromosomal region, is indicated24.
Final considerations
As has been discussed, CHDs are among the most frequent congenital defects, and the patients affected by these anomalies require an early, extensive clinical evaluation, with the goal of establishing either the diagnosis of an isolated CHD or if this is part of a syndromic entity. The genetic aspects of the CHDs presented in this review, underline the importance of the available genomic analyses to establish their etiology. They also remark the importance of having an orchestrated multidisciplinary medical team, who are implicated in the overall medical management of these patients that include the evaluation by a pediatrician, pediatric and/or adult cardiologist, gynecologist, and medical geneticist, among others. Based on the clinical particular characteristics of each patient, the best way of action could be implemented to determine the etiology and to give the best-personalized medical treatment, which includes the genetic assessment.

