











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink1.Introducción
La obesidad tiene un gran impacto en el incremento de los índices de mortalidad de la población mexicana, debido a que se le considera una enfermedad crónica y progresiva. Se caracteriza por una producción excesiva de tejido adiposo, debida a la pérdida del balance entre el consumo y el gasto de energía y está relacionada con el desarrollo de enfermedades cardiovasculares, metabólicas, neurodegenerativas, así como neoplasias.
En el año 2016, la Encuesta Nacional de Salud y Nutrición (ENSANUT) reportó que la prevalencia de obesidad fue mayor en mujeres que en hombres.1 Un año después, los reportes de la Organización para la Cooperación y el Desarrollo Económicos (OCDE) catalogaron a México en el segundo lugar de prevalencia de obesidad y primer lugar en obesidad y sobrepeso, proyectando un aumento del 5% del 2020 al 2030 acorde al patrón de crecimiento observado.2
En los últimos años, se ha investigado cual es la etiología más probable para el desarrollo de esta patología, así como los factores de riesgo que predisponen a la misma. Se considera de naturaleza multifactorial, pero se ha observado que la predisposición genética juega un papel importante, de tal manera que muchas alteraciones se presentan en genes de expresión neuronal, vinculados a la regulación hambre-saciedad.
Las alteraciones más estudiadas dentro de la fisiopatología de la obesidad son las mutaciones de los genes MC4R, BDNF, POMC y LEPR, los cuales guardan una relación muy estrecha en el sistema nervioso central.
La leptina es considerada como una citocina pleiotrópica con actividades en una gran cantidad de tejidos periféricos, que ejerce su acción una vez que interactúa con su receptor (LR). A nivel cerebral, se expresa principalmente en los núcleos arcuato, dorso-medial, ventro-medial y lateral del hipotálamo, en los cuales se activan o inhiben las neuronas como respuesta a la activación del LR (Figura 1). De esta forma, se estimula la síntesis de neuropéptidos anorexigénicos CART y POMC, inhibiendo a los orexigénicos NPY y AgRP.3
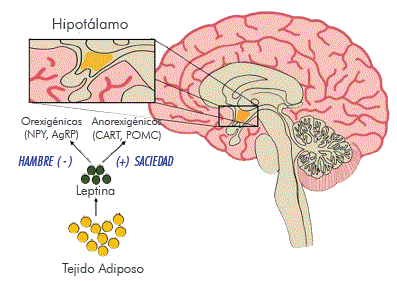
Figura 1 Estimulación de la leptina en el hipotálamo. La leptina inhibe los neuropéptidos orexigénicos (NPY y AgRP), activa a los anorexigénicos (CART y POMC), y produce una señalización de saciedad a nivel neuroendocrino, estimulando la homeostasis, la termogénesis y el balance en el gasto energético.
Estructuralmente, el receptor de la leptina cuenta con seis isoformas, todas estas activan una cascada de señalización a través de las JAK quinasas (Janu Kinasas) para la señalización intracelular. Posteriormente, activan al STAT3, de tal forma que el complejo JAK-STAT3 (Figura 2) constituye un componente muy importante en la señalización de la leptina en el hipotálamo.
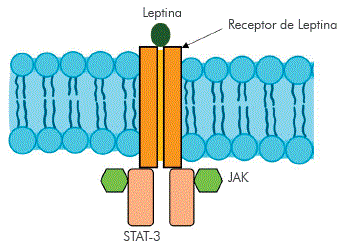
Figura 2 Señalización de la leptina con su receptor. Interacción de la leptina con su receptor, conformando el conjunto JAK-STAT3, componente de dicha señalización.
Debido a que es una hormona polipeptídica producida por los adipocitos, su producción depende de los cambios en el contenido de tejido adiposo y las respuestas adaptativas en el control del balance de energía, siendo sus niveles normales de 5-15 ng/ml en sangre. La obesidad se encuentra influenciada por un incremento de leptina, así como por la resistencia a esta, dado que sus funciones son reducir la ingesta alimentaria generando una sensación de saciedad.
No obstante, la misma obesidad promueve la resistencia a la leptina, esto debido a factores como la alteración en el funcionamiento de su receptor, alteración en las vías neuronales o en tejidos periféricos.4,5 Debido a esta resistencia, los niveles séricos pueden estar por encima de sus rangos normales conforme avanza la enfermedad, lo que lleva a la presentación de hiperfagia en personas obesas.
Por todo lo anterior, estudios realizados en animales mediante la administración de leptina exógena logran el objetivo de reducir la masa corporal en animales normales siendo un determinante importante en el gasto de energía,6 lo cual hace pensar que la administración de leptina como tratamiento de la obesidad es una propuesta tentativa. Por otro lado, la medición de los niveles séricos podría ser un importante predictor para el desarrollo de las posibles comorbilidades a las que predispone.
2.Obesidad
La obesidad es una enfermedad multifactorial compleja, que se desarrolla a partir de la interacción entre el genotipo y el medio ambiente. Nuestro entendimiento de cómo y por qué ocurre es incompleto. Al igual que otras enfermedades, a la obesidad se le ha atribuido un componente genético importante, como puede ser el polimorfismo del gen FTO en la población mexicana.7 Sin embargo, las mutaciones aisladas no logran explicar el rápido aumento y las características epidemiológicas de esta enfermedad, lo que implica una amplia integración de factores sociales, culturales, fisiológicos y metabólicos aunados a los genéticos.7,8
Se han encontrado resultados que comprueban la incrementada mortalidad asociada a la obesidad, particularmente en los casos graves, comparados con el peso corporal normal.9 La obesidad incrementa potencialmente el riesgo de desarrollar enfermedades metabólicas (principalmente la diabetes mellitus tipo II), enfermedades cardiovasculares (hipertensión, infarto agudo al miocardio, evento vascular cerebral, etc.), enfermedades neurodegenerativas (enfermedad de Alzheimer) así como algunos tipos de cáncer (ovario, mama, próstata, hígado, etcétera). La obesidad también se ha asociado con condiciones de salud mental (depresión mayor) así como un aumento del riesgo de complicaciones del binomio madre- hijo (diabetes gestacional, pre-eclampsia, abortos y óbitos).8,10
En materia de salud pública, la diabetes mellitus tipo 2 tiene una estrecha relación con la obesidad, problema que afecta en gran medida a la población. Además de representar un gasto directo importante para la economía de un país, causa costos indirectos por la pérdida de productividad de las personas afectadas y su mortalidad prematura. Los gastos de una persona con diabetes son de 2 a 3 veces superiores a los de una persona que no padece la enfermedad. En América Latina y el Caribe, muchas personas con diabetes tienen acceso limitado a los cuidados de salud. Las consecuencias se traducen también en un mayor costo de cuidados médicos para los individuos, ya que se estima que el gasto en materia de salud de un(a) paciente con obesidad es 42% mayor al de las personas no obesas.8
2.1.Epidemiología de la obesidad
De acuerdo con la Organización Mundial de la Salud (OMS), la prevalencia de obesidad en febrero del 2018 se triplicó a nivel mundial respecto a 1975. Hasta el 2016, más de 1.9 mil millones de adultos padecían sobrepeso/obesidad, de los cuales 34% eran aquellos con obesidad de cualquier grado de severidad.9
Según el reporte de la OCDE del 2017, México se posicionó en el segundo lugar en obesidad y en el primero en sobrepeso/ obesidad, superando a los Estados Unidos, asimismo, se estima que para el 2030 llegará a una prevalencia de 39%.2
En el 2008, 35% de los adultos mayores de 20 años tenían sobrepeso [índice de masa corporal (IMC) mayor a 25]. En ese mismo año, 10% de los hombres y 14% de las mujeres en el mundo eran obesos (IMC superior a 30), comparado con el 5% de hombres y 8% de mujeres en el año de 1980. Un estimado de 205 millones de hombres y 297 millones de mujeres mayores de 20 años son obesos, un total de más de medio billón de adultos alrededor del mundo;10 sin embargo, de acuerdo a la Encuesta de Examinación Nacional de Salud y Nutrición (NHANES), 18.5% de los niños y 39.6% de los adultos tenían obesidad en el periodo de 2015-2016. Estas han sido hasta el momento las cifras más altas documentadas por la NHANES.8 La prevalencia de sobrepeso y obesidad es mayor en el continente americano (62% de sobrepeso en ambos sexos y 26% de obesidad), y menor en la región del Sudeste de Asia (14% de sobrepeso en ambos sexos y 3% de obesidad). En todos los continentes se encontró mayor severidad en mujeres que en hombres.
La presencia de un IMC elevado aumenta con el nivel de ingresos, la diferencia en la prevalencia de sobrepeso en países con altos ingresos económicos es cercana al doble comparada con aquellos con bajos ingresos.10
En el 2012, la prevalencia en la población adulta mexicana se encontraba en el orden del 33%, colocando al país en el primer quintil de la obesidad en América Latina y el Caribe. En un meta-análisis reciente de más de 100 estudios, se encontró que los individuos con o sin sobrepeso comparten riesgos de mortalidad similares y el mayor riesgo de este desenlace está concentrado en aquellos clasificados clínicamente como obesos.11
2.2.Fisiopatología de la obesidad
El abordaje de la fisiopatología de la obesidad puede realizarse con base en cuatro puntos: genético, adiposo, neurológico e inflamatorio.
Si bien se mencionó al inicio el carácter multifactorial de la obesidad, se ha podido observar que hasta 70% del desarrollo de esta enfermedad tiene un patrón de heredabilidad.12 Gracias a los estudios de secuenciación genómica completa (GWAS) se han podido identificar algunos genes que están altamente implicados en el desarrollo de la obesidad con el avance del tiempo, cabe resaltar que gran parte de ellos son genes de expresión neuronal, los cuales se vinculan ampliamente con la regulación de la sensación de hambre-saciedad.
¿l primer gen en ser relacionado con la génesis de la obesidad fue el gen asociado con la masa grasa y la obesidad (FTO por sus siglas en inglés). Este gen se encuentra estrechamente relacionado con el desarrollo de la obesidad en las personas que contienen un determinado polimorfismo de nucleótido único (SNP por sus siglas en inglés).13 Si bien en un inicio las alteraciones atribuibles a los SNP del gen FTO no estaban confinadas al sistema nervioso central (SNC), posteriormente ha comenzado a observarse una asociación con patologías que no están ligadas directamente a la obesidad, como la enfermedad de Alzheimer.14
En la patogénesis de la obesidad, destacan las mutaciones de los genes MC4R, BDNF, POMC y LEPR, los cuales tienen un papel principal en la relación hambre-saciedad en el SNC.
Por otra parte, las comidas con alto contenido calórico generan señales que de forma inmediata estimulan el sobre- consumo de alimentos. Los estudios de imagen han mostrado hiperactivación en la corteza cerebral relacionada con el sentido del gusto (ínsula/opérculo frontal) y las regiones orales somatosensitivas (opérculo rolándico y parietal) en personas obesas -a comparación de los sujetos de peso normal- como respuesta a la anticipación de la ingesta y el consumo de distintos tipos de comida agradable al gusto, además de una hipoactivación en el cuerpo estriado dorsal y una reducción en la densidad de receptores dopaminérgicos D2 estriatales posterior al consumo de dicha comida.
Estos descubrimientos indican la relación que existe entre las anormalidades en la recompensa por comida y el aumento de peso, sugiriendo una mayor ganancia en aquellos que están en contacto con un entorno de comida poco saludable.12 Todo esto conlleva a un exceso de adipocitos que secretan gran cantidad de citocinas que contribuyen a la disfunción vascular en la hipertensión y la dislipidemia, como se manifiesta en la hipercolesterolemia y la trigliceridemia. Estas condiciones eventualmente contribuyen al desarrollo de ateroesclerosis, y, cuando están asociadas a la obesidad, diabetes o la resistencia a la insulina, constituyen el síndrome metabólico.
La grasa almacenada es requerida para la supervivencia durante estados en los que se priva la nutrición; en tiempos de comida prolongada en abundancia, el consumo excesivo de grasa conlleva a un exceso de su almacenamiento, que eventualmente resulta en obesidad. Hay hipótesis que establecen que el almacén de ácidos grasos como triglicéridos dentro de los adipocitos protege contra la toxicidad de los mismos, por otro lado, estos ácidos grasos libres circulantes producen estrés oxidativo diseminándose en todo el cuerpo.
Sin embargo, el almacén excesivo que crea la obesidad lleva eventualmente a la liberación de estos ácidos a partir de la lipolisis, que es estimulada por el sistema nervioso simpático. La liberación de cantidades excesivas de ácidos grasos incita la lipotoxicidad, mientras que los lípidos y sus metabolitos generan estrés al retículo endoplásmico y la mitocondria. Los ácidos grasos liberados de depósitos de triglicéridos también inhiben la lipogénesis, previniendo la eliminación adecuada del nivel sérico de triglicéridos que contribuye a la hipertrigliceridemia.
La liberación de ácidos grasos por la lipasa lipoproteica endotelial a causa del incremento de triglicéridos séricos con elevadas lipoproteínas causa lipotoxicidad que resulta en la disfunción del receptor de insulina.
El consecuente estado de resistencia a la insulina crea una hiperglicemia con gluconeogénesis hepática compensadora, aumentando la producción de glucosa hepática y la hiperglicemia causada por resistencia a la insulina.13
2.3.Adipocitos y adipocinas
La función principal del tejido adiposo es almacenar y dar reserva de energía en la forma de triacilglicerol (triglicéridos). La grasa es almacenada en células especiales, los adipocitos, con funciones específicas. El adipocito tiene un diámetro de aproximadamente 10-12 μm. Después de la acumulación de grasa, su diámetro incrementa 10 veces, eso significa que su volumen aumenta hasta 1,000 veces. El tejido adiposo tiene una enorme plasticidad y capacidad para almacenar energía.
Los adipocitos tienen dos funciones principales: acumular y metabolizar triacilglicerol. El almacenamiento de la grasa depositada en los adipocitos es llevado a cabo por la captura del triacilglicerol por una red capilar de glucosaminoglicanos. Los triacilgliceroles son subsecuentemente hidrolizados por la lipoproteína lipasa. Los ácidos grasos son capturados por los adipocitos adyacentes. La lipoproteína lipasa es sintetizada en el adipocito bajo la influencia de un gran número de hormonas, siendo la insulina y el cortisol las principales.14
Los adipocitos, que representan más de un billón de células, no solo almacenan triglicéridos en depósitos de grasa en varios sitios corporales para proveer energía, también constituyen el tejido endocrino más grande, en constante comunicación con otros tejidos por distintas sustancias, como las proteo-hormonas lectina, adiponectina y visfatina. Junto con la insulina, estas proteo-hormonas ayudan a regular la grasa corporal. Otros grupos de genes que contribuyen a las adipocinas de los adipocitos incluyen las citocinas, factores de crecimiento y proteínas del complemento.
La dislipidemia, la hipertensión y la aterogénesis son condiciones que, junto con la resistencia a la insulina, están asociadas a la obesidad. Asimismo, están influenciadas por la secreción de diversas adipocinas inflamatorias, particularmente por los tejidos adiposos blancos en los depósitos de grasa viscerales. Algunas adipocinas específicas aumentan el tono vasomotor por la secreción de renina, angiotensinógeno y angiotensina II, que son similares a aquellos en el sistema renina angiotensina, pero cuando son secretados por los adipocitos establecen la hipertensión en las personas obesas; la secreción del factor de necrosis tumoral estimula la inflamación.13
3.La leptina y su relación con la obesidad
Una gran cantidad de hormonas periféricas participa en el control del apetito, la ingesta de comida, la recompensa alimenticia, o la adicción, por parte del sistema nervioso central. Tanto la comida agradable al gusto como las drogas activan la vía dopaminérgica mesolímbica involucrada en el sistema de recompensa, que es esencial para la regulación de la adicción en humanos y animales.
El hambre y las señales del tejido adiposo (leptina), el páncreas (insulina), y el sistema gastrointestinal (colecistoquinina, péptido parecido al glucagón-1, péptido YY3-36, y la ghrelina) aportan parte de la información que regula el estado de energía a través del eje neurohormonal intestino-cerebro, primariamente hacia el hipotálamo y el tallo encefálico, y pueden interactuar directa o indirectamente con las vías dopaminérgicas del mesencéfalo para impactar la alimentación.12
3.1.La leptina
La leptina juega un rol central en el control del peso corporal y la homeostasis, pero es una citosina pleiotrópica, con actividades en una gran cantidad de tejidos periféricos. Esta hormona es conocida por participar en un amplio rango de funciones biológicas, incluyendo inmunidad innata e inmunidad adaptativa,15 reproducción16 y formación de hueso.17
La leptina cruza la barrera hematoencefálica por vía de un sistema de transporte saturable y comunica el estado metabólico de la periferia a los centros reguladores del hipotálamo. Una vez que se une a su receptor, la leptina inhibe los neuropéptidos que estimulan el apetito (NPU, AgRP) mientras estimula a la hormona estimulante alfa- melanocítica y la hormona liberadora de corticotropina.12 El gen de la leptina está localizado en el brazo largo del cromosoma 7 (7q31.3) y contiene 3 exones y 2 intrones.18
La leptina es una proteína no glicosilada de 16 kDa y de 146 aminoácidos. La leptina humana tiene expuestos dos residuos de triptófano. Adopta la estructura típica de una citosina de 4 hélices (A, B, C y D), en una forma arriba-arriba-abajo-abajo. Los residuos del extremo C terminal de la hélice D adoptan una estructura 3-10. Dos vueltas largas AB y CD conectan las hélices paralelas, mientras que las hélices antiparalelas B y C están conectadas por una vuelta corta BC. La leptina contiene 2 residuos de cisteína (C96 en la vuelta CD y C146 en el residuo C terminal), este puente disulfuro es esencial para la estabilidad estructural y su actividad biológica.19
La leptina es miembro de una familia de citosinas de cadena larga, sin embargo, sus hélices son en promedio una o dos vueltas más cortas, como consecuencia, posee la rama más corta de dicha familia.
3.2.Receptor de leptina
Estructuralmente, el LR puede ser clasificado como un receptor de citosina de clase I. Esta familia consiste en un receptor de membrana único que abarca los receptores marcados por la presencia de uno o más receptores de citocinas de dominios homólogos. Las cualidades extracelulares pueden incluir dominios tipo inmunoglobulina y fibronectina tipo III.
Todas las isoformas del LR utilizan las JAK quinasas para la señalización intracelular (Figura 2). Hasta ahora se conocen 6 isoformas del receptor de leptina: LRa, LRb, LRc, LRd, LRe y LRf. Todas estas isoformas, excepto la LRe, tienen un dominio transmembranal extracelular idéntico, pero difieren en la longitud de la cola intracelular. LRb, también referida como la forma larga del LR, tiene 1162 residuos, con un dominio intracelular de 302; se piensa que es la única isoforma que emplea la señalización con el complejo JAK/STAT. Esta isoforma está altamente expresada en núcleos específicos del hipotálamo, donde lleva a cabo su función en la regulación del peso corporal.
LRa, LRc, LRd y LRf tienen solamente una cola intracelular corta (30-40 residuos) y un único dominio C terminal. Su rol fisiológico preciso sigue siendo poco claro, pero altos niveles de expresión de la LRa y el LRc en los plexos coroideos y en la microvasculatura cerebral pueden sugerir un rol importante en el transporte de leptina a través de la barrera hematoencefálica.20
Inicialmente descubierto como una mutante defectuosa,21 LRe en los ratones es una variante soluble, que es directamente secretada en el torrente sanguíneo. En los humanos, un fragmento similar de LR es generado por un ecto-dominio proteolítico (metaloproteasa 10 y 17)22 y modula la biodisponibilidad de la leptina.
Existen distintas isoformas del LR, conocidas como isoformas cortas y largas. El gen del LR está localizado en el brazo corto del cromosoma 1 (1p31) y contiene 20 exones.23
El LR contiene 6 dominios extracelulares: un Dominio N terminal (NTD) de función y estructura no definidas, un primer dominio CRH (CRH1), un IGD, un segundo CRH (CRH2) y dos dominios proximales FNIII. Se desconoce la función del NTD. El CRH1 no es necesario para la señalización del LR,24 en contraste, la señalización es completamente abolida por la eliminación de cualquiera de los otros cuatro dominios. El dominio CRH2 es el único dominio de alta afinidad para la leptina, a diferencia de los otros dominios que no muestran un sitio especifico de la unión de la leptina.
4.La leptina y la obesidad
La leptina es una hormona polipeptídica que es producida por los adipocitos en proporción a su contenido de triglicéridos, y enlaza los cambios en el almacén de energía corporal (grasa) con las respuestas adaptativas en el control central del balance de energía. Por unión y activación de la forma larga de su receptor (LR-B) en el cerebro, la leptina disminuye el hambre mientras aumenta el gasto de energía.25
La leptina circula a niveles de 5 a 15 ng/ml, su expresión se ve aumentada por la ingesta de alimentos, insulina, glucocorticoides, endotoxinas, y citocinas, y disminuida por la testosterona, hormona tiroidea, y la exposición a bajas temperaturas. En el corazón se observa un aumento en la expresión de leptina en la reperfusión después de la isquemia.26 Se ha demostrado que su papel principal es evitar y responder ante las reducciones de la grasa corporal.27
En un estudio reciente, se ha observado que la falta de leptina en ratones puede causar obesidad severa por el aumento en la ingesta de comida y el menor gasto de energía;28 estudios similares lo han comprobado en seres humanos,29 dichos efectos pueden ser reversibles tras la administración de leptina.30
La obesidad no está influenciada únicamente por un déficit de leptina, sino también por la resistencia a la misma. Dado que la presencia de leptina reduce la ingesta de alimentos y el peso corporal, pueden observarse elevados niveles de leptina en las personas obesas con resistencia a la leptina. Los efectos de la resistencia a la leptina son reversibles. Por ejemplo, si el contenido de grasa de los ratones obesos se reduce, los ratones recuperan la sensibilidad a la leptina y el control glicémico. Se cree que el aumento de sensibilidad a la leptina en los circuitos de melanocortina tiene influencia sobre la resistencia a esta.
Algunos estudios en animales han encontrado que la resistencia a la leptina inducida por la dieta ocurre en distintas etapas. En la primera etapa, ratones con una dieta alta en grasa presentaron sensibilidad a la leptina exógena.
En la segunda etapa, al disminuir el consumo de comida se produjo un aumento en la producción y sensibilidad a leptina. En la etapa final, se aumentó la ingesta de comida y se redujo la sensibilidad central a la leptina (Figura 3).
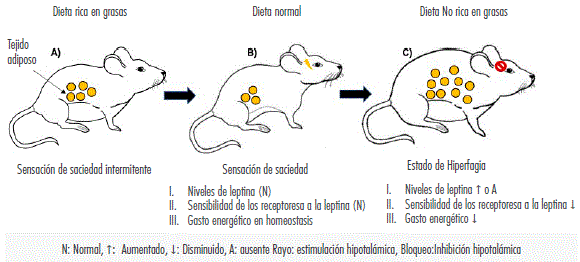
Figura 3 Obesidad secundaria a ausencia de leptina o por dieta rica en grasas y sus estados. A) Ratones con dieta alta en grasa + administración de leptina exógena (sensibilidad conservada intermitente a la inducción). B)Reducción en el consumo de alimentos, así como del peso, aumento en la producción y sensibilidad a la leptina central. C) Aumento en la ingesta de alimentos, reducción de la sensibilidad de leptina central, disminución del gasto energético = Ratón con obesidad.
La resistencia a la leptina causada por una alta ingesta de grasa resulta en un defecto en el acceso a distintos sitios de acción en el hipotálamo, lo cual disminuye significativamente la habilidad de la leptina periférica para activar la señalización hipotalámica. La resistencia es también causada por un defecto en la señalización intracelular en neuronas hipotalámicas que responden a leptina.
La administración aguda de leptina exógena reduce el hambre y la masa corporal en los animales, y es un determinante importante de gasto de energía. Estas observaciones establecen que la deficiencia de leptina es una llave reguladora de respuestas metabólicas y neuroendocrinas a estados caracterizados por balance negativo de energía y pérdida de peso, razón por la cual el mantenimiento de la pérdida de peso es poco exitoso.6
La resistencia a la leptina ocurre también como una respuesta fisiológica adaptativa para permitir cambios plásticos en mecanismos homeostáticos, permitiendo alteraciones repetidas y reversibles en el peso corporal en ciertas situaciones.31 En años recientes se ha presenciado un incremento dramático en el número de modelos genéticos de ratones con obesidad, por lo que medir la sensibilidad a la leptina se ha convertido en un proceso de rutina en los esfuerzos para investigar los mecanismos de la obesidad.
La mayoría de las personas obesas (Figura 4), exhiben valores elevados de leptina circulante, asociados con su nivel de tejido adiposo.32
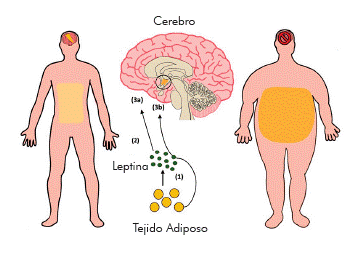
Figura 4 Regulación del apetito mediado por la leptina. 1.Producción dela hormona leptina a partir del tejido adiposo (adipocitos). 2. Liberación a torrente sanguíneo para estimulación o inhibición hipotalámica. 3a. Síntesis de neuropéptidos anorexigénicos e inhibición de los orexigénicos. 3b. Resistencia a la leptina que condiciona hiperleptinemia o almacenamiento excesivo de tejido adiposo, producción pobre de leptina (ambos condicionan gasto energético bajo e hiperfagia).
La obesidad promueve varias vías de resistencia a la leptina, por lo que la acción de la leptina puede estar comprometida en animales obesos. Asimismo, se consideran algunas clases de obesidad genética relacionadas a la leptina4
Alteraciones en LR-B o su señalización.5
Alteraciones en las vías neurales involucradas en la acción de la leptina.
Alteraciones en los tejidos periféricos que operan independientemente del consumo de alimentos.
Alteraciones en las vías del sistema nervioso central sin un enlace claro con la acción de la leptina.
La leptina también se ha asociado a ateroesclerosis, el engrosamiento de la íntima y la túnica media; se cree que es un marcador de las etapas iniciales de la ateroesclerosis, antes de que los síntomas inicien. Investigadores han mostrado que la concentración sérica de leptina se relaciona independiente y de manera positiva con el engrosamiento de la íntima- media de la arteria carótida común.33 De igual modo -como se comentó previamente- dado su papel en la promoción de un estado proinflamatorio persistente acompañado de la presencia de obesidad, esto promueve la estimulación de los receptores de leptina tipo B, la oxidación de ácidos grasos a nivel cardiaco, el aumento del consumo de oxígeno por parte del miocardio, así como el aumento de especies reactivas de oxígeno, entre otros. Esto promoverá la disfunción miocárdica, lipotoxicidad, así como un manejo inadecuado de calcio dentro de los miocitos así como una fibrosis a largo plazo, lo cual está vinculado con la fisiopatología de la cardiomiopatía diabética y disfunción por fibrosis.34
5.Regulación dopaminérgica de la conducta alimentaria
Los mecanismos neuroquímicos que controlan el peso corporal y el estado de ánimo son similares.35 La ingesta de alimentos es una conducta dirigida por la motivación de supervivencia, por lo que su regulación involucra al sistema de neurotransmisión dopaminérgica, el cual también se encuentra directamente involucrado en la fisiopatología de las adicciones, además de los mecanismos del reforzamiento y la recompensa.36 Estos sistemas pueden encontrarse alterados en personas que padecen trastornos de la alimentción como anorexia nervosa y bulimia nervosa.37
Algunos estudios han mostrado que la leptina puede modular la recompensa generada por la ingesta de comida con alto contenido de grasas.36 Estos efectos pueden estar influenciados por la presencia de polimorfismos de nucleótido único en el gen que codifica para la leptina.38 Esta convergencia entre los mecanismos de señalización permite que la leptina se encuentre involucrada en la presentación de trastornos neuropsiquiátricos en personas que padecen obesidad.39
Las neuronas del área tegmental ventral (VTA), origen de la vía dopaminérgica mesolímbica, expresan receptores para leptina,40 cuya activación aumenta la expresión de tirosina hidroxilasa y el contenido de dopamina en esas neuronas, pero reduce la liberación de dopamina en el núcleo accumbens.41 Dichas neuronas responden a los estímulos que predicen la obtención de alimentos (condicionamiento). Las proyecciones del núcleo lateral del hipotálamo hacia la VTA estimulan la ingesta de alimentos a través de las proyecciones de la VTA hacia el núcleo accumbens.40
La conducta alimentaria también se encuentra regulada por otras vías de señalización, como el eje intestino-cerebro,42 mediado, al menos en parte, por el nervio vago.43 El intestino puede secretar diversos péptidos que contribuyen a la regulación del apetito actuando al nivel del VTA y el núcleo accumbens.42 Además de los péptidos intestinales, su microbioma también podría estar involucrado en la fisiopatología de la obesidad.44
6.Conclusiones
La leptina juega un papel muy importante en la etiología de la obesidad, debido a su implicación en los diversos procesos fisiológicos y fisiopatológicos propios del organismo. Al ser una hormona relacionada con la saciedad, sus alteraciones tienen implicaciones a nivel neuroendocrino, esto debido a un defecto en su producción o una alteración en su captación que condicionan un estado de apetito insaciable e hiperfagia. En los últimos años, se ha recalcado la importancia de la predisposición genética para el desarrollo del sobrepeso y la obesidad; asimismo, se ha relacionado la producción de esta hormona con el porcentaje de tejido adiposo en el organismo, encontrando que, a mayor tejido graso, habrá mayor resistencia a la captación de leptina. A partir de esto, se ha propuesto medir los niveles de esta hormona como un marcador de predisposición para el desarrollo de ateroesclerosis, alteraciones en la estructura vascular, así como el desarrollo de comorbilidades. La administración de leptina exógena parece ser una solución innovadora para la resolución de la obesidad en aquellos candidatos que padezcan deficiencia de esta, promoviendo una disminución del apetito y la regulación del peso corporal.
Debido a la importancia que juega la leptina en el desarrollo de la obesidad, es importante recalcar que las investigaciones realizadas son clave para comprender los mecanismos y las señalizaciones neuroendocrinas que condiciona la ausencia, resistencia u homeostasis en la relación tejido adiposo-leptina-hipotalamo. Cada vez estamos más cerca de entender la relación genómica con los factores predisponentes para el desarrollo del sobrepeso y obesidad. La ausencia de ciertos genes condiciona la deficiencia de esta hormona o la resistencia de su receptor.
Aún queda mucho por analizar de esta patología, por lo cual sería de suma importancia invertir en investigaciones y modelos experimentales para una mayor compresión de dichos procesos fisiopatológicos, así como la búsqueda de una solución para la patología.

