











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
Antecedentes
Las reacciones adversas a medicamentos se definen como cualquier reacción perjudicial, indeseada o no prevista, provocada por algún fármaco. Los efectos pueden ocurrir con las dosis administradas para la profilaxis, el diagnostico o tratamiento de alguna enfermedad.1
Las respuestas se dividen en 1) tipo A, relacionadas con la acción del medicamento, influenciadas por la dosis, farmacocinética, condiciones médicas y posibles interacciones; y 2) tipo B, que se encuentran menos definidas e implican respuestas del sistema inmunológico, y la mayor parte se consideran con hipersensibilidad a medicamentos, representando aproximadamente el 15% de todas las reacciones adversas a medicamentos.2
En términos médicos, las respuestas de hipersensibilidad se clasifican en inmediatas (manifestándose de 1 a 6 horas luego de la exposición) y tardías o no inmediatas (que se originan 6 horas después de la exposición).3 Las reacciones alérgicas clásicas se separan en cuatro tipos, siguiendo el esquema de Coombs y Gell. Las reacciones de tipo I, II y III están mediadas por anticuerpos, mientras que las reacciones de tipo IV por células.4
Las reacciones de tipo I se caracterizan por un inicio agudo e implican la activación de mastocitos y basófilos, y están mediadas por anticuerpos IgE. Las reacciones de tipo II inician días después de la exposición e implican destrucción celular mediada por anticuerpos IgG o IgM. Las reacciones tipo III tienen un inicio retardado y resultan de la formación de anticuerpos-complejos que se depositan en los tejidos y provocan la activación del complemento, con subsiguiente daño tisular.3 Figura 1
Las reacciones tipo IV son retardadas y están mediadas por células T activadas, ocurren entre 48 a 72 horas de la exposición. las manifestaciones clínicas pueden variar desde una erupción cutánea leve (Figura 2) hasta enfermedades graves (Figura 3): síndrome de Stevens-Johnson-necrólisis epidérmica tóxica (SJS/NET). Las reacciones adversas mediadas por células T están fuertemente relacionadas con la portación de alelos de riesgo de antígenos leucocitarios humanos (HLA) particulares.3,5
El objetivo de este estudio fue describir las reacciones de hipersensibilidad tardía provocada por fármacos, su epidemiologia, manifestaciones clínicas, diagnóstico, tratamiento y pronóstico.
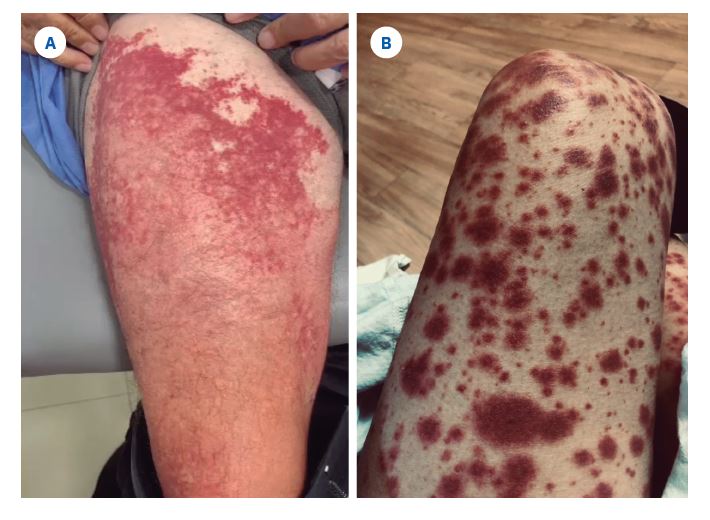
Figura 1 Reacciones de hipersensibilidad tipo III (vasculitis). A) Lesiones de vasculitis cutánea con aparición de eritema extenso, livedo racemosa y múltiples petequias en extremidad inferior. B) Lesiones de vasculitis cutánea caracterizada por múltiples pápulas purpúricas en extremidad inferior.
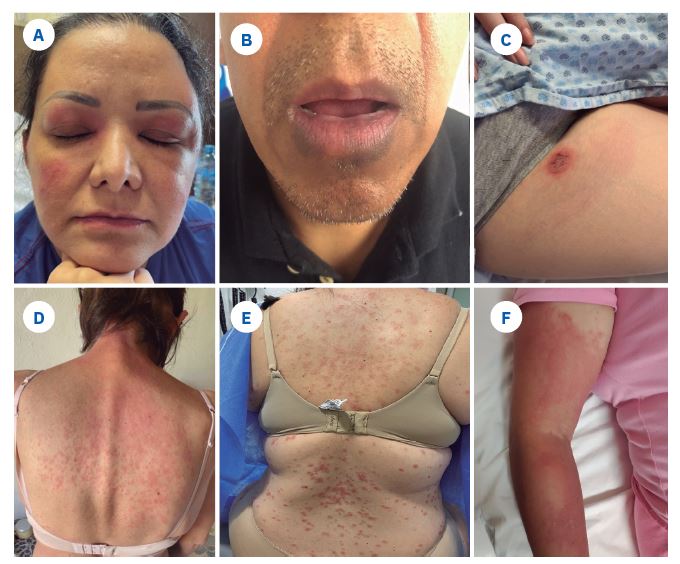
Figura 2 Reacciones de hipersensibilidad tipo IV (lesiones leves). A) Dermatitis de contacto sistémica en los parpados de forma bilateral, y las mejillas. B) Eritema fijo pigmentado, con mancha solitaria, bien delimitada, eritematosa a violácea, redonda u ovalada, con centro oscuro. C) Exantema fijo ampolloso, manifestado con ampolla-vesícula, dejando una úlcera superficial. D) Exantema conformado por máculas y pápulas eritematosas, polimorfas y confluentes en el tronco y las extremidades. E) Lesiones de reacción liquenoide, caracterizada por placas planas, poligonales, que varían en tamaño, pruriginosas; en el tronco y las extremidades. F) Lesión de SDRIFE (exantema intertriginoso y flexural simétrico relacionado con fármacos), distinguida por eritema simétrico que afecta los pliegues, en la extremidad superior.
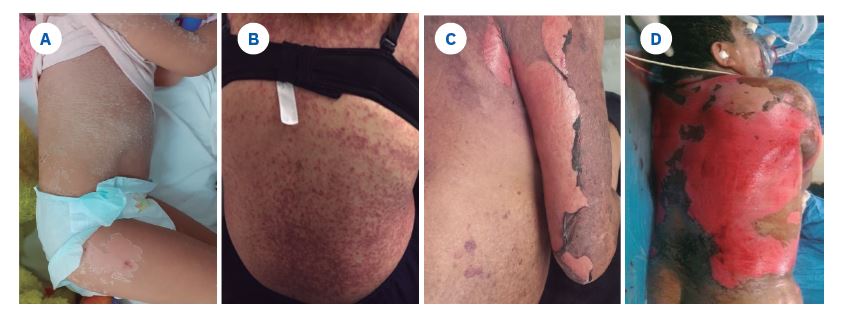
Figura 3 Reacciones de hipersensibilidad tipo IV (lesiones graves). A) Lesión de AGEP (Pustulosis exantemática aguda generalizada), caracterizada por pústulas pequeñas, estériles y no foliculares en una base eritematosa, distribuidas en el tronco y las extremidades. B) Lesión por síndrome DRESS (Síndrome de Hipersensibilidad a Medicamentos con Eosinofilia y Síntomas Sistémicos), manifestado por exantema maculopapular característico, principalmente en el tronco y las extremidades. C) Síndrome de Stevens-Johnson (SJS), con lesiones eritematosas o en diana atípicas en el tronco y la extremidad superior que progresan y confluyen con centro obscuro, además de signo de Nikolsky positivo y denudación cutánea. D) Lesión de NET (Necrólisis Epidérmica Tóxica) con afectación de las mucosas y la piel, en el tronco, con ampollas flácidas y signo de Nikolsky positivo, con denudación cutánea.
Métodos
La búsqueda de la información se llevó a cabo en las principales bases de datos, en relación con las reacciones de hipersensibilidad tardía a medicamentos. La búsqueda se limitó a artículos publicados entre 2013 y 2023, en idioma inglés y español.
Dermatitis de contacto y dermatitis de contacto sistémica
La dermatitis alérgica de contacto es una enfermedad inflamatoria de la piel, que resultan del contacto de un antígeno ofensivo con ésta. La dermatitis de contacto sistémica se caracteriza por erupción cutánea luego de la exposición sistémica (transcutánea, transmucosa, oral, parenteral, intraarticular, intravesical, inhalatoria e implantes) a alérgenos, después de la sensibilización cutáneamucosa previa.6
Fisiopatología
Son reacciones de hipersensibilidad retardada tipo IV, mediada por células T, que ocurren aproximadamente después de 48 horas de la exposición al alérgeno.7
Epidemiología
Entre el 15-20% de la población mundial tiene dermatitis de contacto, mínimo a un alérgeno; sin embargo, los datos epidemiológicos son difíciles de estimar, porque ambos tipos de dermatitis comprenden diversas manifestaciones clínicas. Es más común en mujeres y en personas de la tercera edad.7
Cuadro clínico
La dermatitis alérgica de contacto se manifiesta con pápulas pruriginosas, eritematosas, edematosas, y vesículas en el sitio de contacto o en sitios distantes por autoinoculación; como exacerbación de la dermatitis o retraso en la cicatrización de las heridas.8
La dermatitis alérgica de contacto ocupacional suele ocurrir en trabajadores sanitarios, debido a la manipulación de medicamentos, lavado frecuente de manos y uso prolongado de guantes.9 La dermatitis transmitida por el aire puede afectar áreas expuestas (cara, cuello, manos, muñecas, axilas) o no expuestas (por partículas atrapadas en la ropa).10
Las manifestaciones en pacientes con dermatitis alérgica de contacto sistémica incluyen eczema, o su reactivación en las pruebas de parche: erupción maculopapular, ronchas, eritema multiforme, dermatitis fotoalérgica y ocasionalmente síntomas sistémicos.11 La manifestación habitual es eritema en los glúteos, muslos y pliegues axilares, y se denomina “síndrome del babuino”.12
El Cuadro 1 muestra los fármacos más frecuentes relacionados con la dermatitis de contacto.
Diagnóstico Para establecer el diagnóstico dermatitis de contacto se realizan pruebas de parche, administrando el ingrediente activo y excipientes sospechosos.
Cuadro 1 Reacciones adversas a medicamentos, características y agentes farmacológicos desencadenantes.
| Reacción adversa a medicamentos | Mecanismo | Célula efectora | Fármacos implicados |
|---|---|---|---|
| Dermatitis por contacto | Hipersensibilidad tipo IV | Células T |
|
| Erupción fija pigmentada por fármacos | Hipersensibilidad tipo IV | Células T | |
| Eritema multiforme | Hipersensibilidad tipo IV | Células T |
|
| Exantema maculopapular | Hipersensibilidad tipo IV | Células T | |
| Exantema intertriginoso y flexural simétrico relacionado con fármacos | Hipersensibilidad tipo IV | Células T |
|
| Reacción liquenoide a fármacos | Hipersensibilidad de tipo IVc | Células T |
|
| Pustulosis exantemática aguda generalizada | Hipersensibilidad de tipo IVd | Células T |
|
| Síndrome DRESS | Hipersensibilidad de tipo IVd | Células T |
|
| Síndrome de Stevens-Johnson / necrólisis epidérmica tóxica | Hipersensibilidad de tipo IVc | Células T |
|
| Enfermedad del suero / Reacción de Arthus | Hipersensibilidad tipo III | Depósito de complejos inmunes |
|
| Vasculitis inducida por medicamentos | Hipersensibilidad tipo III | Depósito de complejos inmunes |
|
| Citopenias autoinmunes | Hipersensibilidad tipo II | Anticuerpos IgG o IgM |
|
*Adaptado de 7,8,16,19,21,23,25,26,29,54,65,70-73,83-90. Consulte la lista de referencias para obtener detalles completos de las fuentes.
Tratamiento y pronóstico
El padecimiento suele desaparecer días después de suspender los alérgenos, mediante la administración de corticosteroides tópicos para la inflamación cutánea leve y el prurito, y corticosteroides sistémicos para casos más graves.6 Después de identificaron los alérgenos y sus reactividades cruzadas, debe instruirse a la lectura de etiquetas de medicamentos tópicos.8
Eritema fijo pigmentado y eritema fijo pigmentado buloso (ampolloso)
La erupción fija pigmentada provocada por fármacos es la recurrencia de una erupción en el mismo sitio cada vez que se ingiere algún medicamento. Luego de cada exposición las lesiones pueden aumentar de tamaño y cantidad de sitios afectados.13,14 El tiempo entre la exposición y la aparición del eritema varía de unas cuantas horas hasta dos semanas, con un promedio de 48 h.14-17
Fisiopatología
Son reacciones de hipersensibilidad tipo IV, mediadas por células T CD8+, que aparecen en las primeras 24 horas de la ingesta del medicamento y posterior a la curación del brote pueden permanecer inactivas durante años en el sitio de lesión.18
Epidemiología
El eritema fijo pigmentado afecta en igual proporción a hombres y mujeres, y la incidencia varía de un continente; sin embargo, el promedio de incidencia se estima en 11 y 30%.15 Otros estudios muestran distribución de las lesiones según el género, por ejemplo en el 89% de las mujeres afecta las extremidades y en el 90% de los hombres los genitales.15,19
Cuadro clínico
En pacientes con eritema fijo pigmentado no ampolloso la manifestación típica es una mancha solitaria, bien delimitada, eritematosa a violácea, redonda u ovalada, con un centro oscuro. Después de desaparecer la inflamación aguda, puede haber hiperpigmentación posinflamatoria, que perdura semanas a meses.
En sujetos con eritema fijo pigmentado ampolloso las lesiones pueden expresarse con ampollas, vesículas y/o bulas, que se rompen con facilidad, dejando erosiones o úlceras superficiales.14,17 En algunos casos puede haber erupción extensa de las ampollas, además de los parches característicos del eritema fijo pigmentado, alteración conocida como erupción ampollosa fija generalizada.14
Algunos autores consideran afectación mínima del 10% del área de la superficie corporal y tres de seis sitios anatómicos diferentes (específicamente la cabeza y el cuello, tronco anterior, espalda, extremidades superiores, inferiores y genitales).20,21
Los fármacos más frecuentes relacionados con el eritema fijo pigmentado se enlistan en el Cuadro 1.
Diagnóstico
El diagnóstico se establece por las manifestaciones clínicas; en caso de duda puede obtenerse una biopsia de la lesión y el resultado histopatológico suele mostrar datos de dermatitis de interfase vacuolar con infiltración perivascular superficial y profunda de eosinófilos y linfocitos. Pueden visualizarse queratinocitos necróticos individuales dispersos por toda la epidermis, y es típica la incontinencia pigmentaria.22 La prueba de parche es una opción, pero la sensibilidad puede no ser adecuada y variable.23,24
Tratamiento y pronóstico
Evitar el uso del medicamento implicado e identificado es el tratamiento ideal en ambos casos tipos de eritema fijo. Pueden indicarse ciclos cortos de corticosteroides tópicos y sistémicos, incluso se han reportado resultados positivos y alentadores con ciclosporina en adultos y niños, pero las dosis deberán individualizarse en cada caso.
El pronóstico regularmente es bueno, con curación luego de algunas semanas.13-15
Eritema multiforme
Es una enfermedad aguda y de curación espontánea, relacionada con reacciones de hipersensibilidad a virus (90%) y herpes simple (70-80%).25-28 Mycoplasma pneumoniae es el segundo agente casal implicado,29,30 mientras que los fármacos son responsables de menos del 10% de los casos.29
Fisiopatología
El eritema multiforme generado por fármacos se asocia con TNF-a, perforina y granzima B, que causan destrucción epidérmica en pacientes con enfermedades con participación de células T CD8 efectoras.26
Epidemiología
La incidencia es menor del 1%,29 afecta a adultos jóvenes de 20 a 40 años, sobre todo mujeres (1.5:1.0). La prevalencia es menor del 1% y no hay asociación con la raza.26
Cuadro clínico
Aparecen lesiones eritematosas en diana, caracterizadas por anillos menores de 3 cm,25-27 de bordes definidos, y tres zonas diferenciadas; dos anillos concéntricos con cambio de color que rodean una zona circular central oscura, manifestando daño en la epidermis en forma de bulla o costra.25,27 Distribución simétrica en las extremidades (superficies dorsales de manos, pies, codos y rodillas) y con propagación centrípeta.26,28,29,30
Se subdivide en eritema multiforme menor (≤ 1 sitio mucoso) y mayor (≥ 2 sitios mucosos).25,26,28
Los síntomas prodrómicos (fiebre, malestar general, cefalea, tos, rinitis, dolor de garganta, mialgia, artralgia, náuseas) aparecen 7 a 14 días antes de la lesión cutánea.26,29 Afecta la mucosa oral en más del 70% de los pacientes.>26 Suele curarse de forma adecuada, sin complicaciones.>29
Los fármacos asociados con esta alteración se muestran el Cuadro 1.
Diagnóstico
El diagnóstico se establece por las manifestaciones clínicas. A diferencia de la urticaria, donde aparece edema de la capa epidermoide, el eritema multiforme no genera edemas, además de las distintas lesiones elementales.29,31
Exantema maculopapular
El exantema morbiliforme también se conoce como reacción exantemática a fármacos o exantema maculopapular secundario a fármacos.
Fisiopatología
Los polimorfismos genéticos de antígenos leucocitarios humanos son responsables de las reacciones adversas cutáneas a medicamentos; además, las infecciones virales subclínicas, pacientes con inmunosupresión o enfermedad autoinmune y la polifarmacia incrementan el riesgo de exantema morbiliforme. Es una reacción de hipersensibilidad tipo IV.32-35
Epidemiología
Afecta entre el 2 y 8% de los pacientes hospitalizados; la incidencia reportada es del 2% en la población general, con predominancia entre los 21-39 años, pero puede aparecer a cualquier edad. La proporción hombre-mujer es de 1:0.9. La medicación por vía oral generar el 79.6% de las reacciones.
Cuadro clínico
Es una dermatosis difusa, de aparición repentina y evolución gradual, constituida por máculas y pápulas eritematosas, en ocasiones polimorfa y confluente, que inicia en el tronco y las extremidades, sin afectación de la cara, las palmas y plantas. Los signos y síntomas más frecuentes son prurito, discreta eosinofilia y elevación de la temperatura corporal, incluso puede afectar las mucosas. El porcentaje de afectación de la superficie corporal es menor del 10%. El periodo de latencia oscila entre 5 y 14 días después del inicio del fármaco y algunas veces 1 a 2 días después de suspenderlo.
Los fármacos más frecuentemente relacionados con el exantema maculopapualr se exhiben el Cuadro 1.
Diagnóstico
El diagnóstico se establece por sospecha clínica; lo apoya la aparición del exantema después de la administración de algún fármaco y su posterior curación al suspenderlo. En caso de duda diagnóstica solo deben efectuarse pruebas de laboratorio. La biopsia es necesaria. La histopatología es inespecífica. Las infecciones virales son causa importante de exantema maculopapular, especialmente en niños, por lo que este aspecto debe considerarse en el diagnóstico diferencial.
Exantema intertriginoso y flexural simétrico relacionado con fármacos (SDRIFE)
Esta alteración es secundaria a la exposición de un fármaco administrado por vía sistémica, en dosis inicial o repetida, excepto los alergenos de contacto clásicos.
Fisiopatología
Es una reacción de hipersensibilidad tipo IV retardada mediada por células T, con aumento de la selectina CD26P en las células endoteliales y queratinocitos, implicada en el reclutamiento de linfocitos Th1. La distribución puede originarse por un fenómeno físico, secundario a la estimulación mecánica, en áreas previamente afectadas por cierto tipo de dermatosis intertriginosa.38,39
Epidemiología
Suele afectar a pacientes de mediana edad, con predilección en los hombres (proporción hombre-mujer 3:1), contrastante con otras enfermedades medicamentosas maculopapulares que suelen ser frecuentes en mujeres.
Cuadro clínico
Se caracteriza por eritema bien delimitado del área glútea o perianal, o eritema de forma V en el área inguinal o perigenital, con afectación de al menos un pliegue flexural adicional, simetría de las zonas afectadas, ausencia de síntomas y signos sistémicos.
Los fármacos más frecuentes relacionados con el exantema intertriginoso y flexural simétrico aparecen en el Cuadro 1.
Reacción liquenoide a fármacos
La reacción liquenoide a fármacos y el liquen plano cutáneo son similares en cuanto a sus características clínicas e histológicas, y ocurre como una reacción adversa a diversos medicamentos.40-42
Fisiopatología
La fisiopatología implica, principalmente, al linfocito T CD8+, y se trata de una hipersensibilidad tipo IVc. Al principio, las citocinas Th1 secretadas por las células T CD4 helper (hipersensibilidad tipo IVa) atacan a los queratinocitos basales, causando una disrupción en la membrana basal. Esto permite la infiltración de linfocitos T CD8+ y genera un ciclo destructivo de queratinocitos por apoptosis, con mayor liberación de granzima B, en comparación con lo que ocurre en pacientes con liquen plano común.41,42
Epidemiología
El liquen plano cutáneo afecta entre el 0.5 al 1% de la población, mientras que la reacción liquenoide a fármacos es excepcional.40,41 Afecta a adultos mayores, sin predilección del género o etnia.40
Cuadro clínico
El lapso entre la exposición al medicamento y la dermatosis varía de una semana a meses o años. Se caracteriza por pápulas o placas pruriginosas, planas y poligonales, generalmente simétricas en el tronco y las extremidades expuestas al sol. A diferencia del liquen plano clásico, no aparecen estrías de Wickham; es polimorfo, con descamación similar a la psoriasis o el eczema, tiende a hiperpigmentarse y afecta en menor proporción las mucosas y uñas, los liegues de flexión y los genitales.40,41
en el Cuadro 1 se enumeran los fármacos más frecuentemente relacionados con la reacción liquenoide a fármacos y el liquen plano.40-42
Diagnóstico
La historia clínica detallada, con mención de los fármacos desencadenantes es decisiva en caso de sospecha clínica. La prueba de parche puede dar falsos negativos y muestra una reacción liquenoide o eccematiforme. La biopsia revela dermatitis liquenoide en la interfase, cuerpos de Civatte, infiltrado linfocítico, incontinencia pigmentaria y eosinófilos en la dermis, lo que diferencia del liquen plano común y sugiere una reacción liquenoide por fármacos.40-42
Tratamiento
Es importante suspender el medicamento sospechoso. Los corticosteroides tópicos de alta potencia aceleran la curación; también pueden prescribirse antihistamínicos.40-42 En pacientes con reacciones generalizadas se recomiendan corticosteroides sistémicos. Otros protocolos incluyen retinoides sistémicos o ciclosporina; sulfasalazina y fototerapia UVB de banda ancha-banda estrecha, o psoraleno y UVA. Inhibidores de la calcineurina, hidroxicloroquina, azatioprina, metotrexato, micofenolato mofetilo o productos biológicos dirigidos a IL-12/23. Algunos estudios valúan el tratamiento con anticuerpos monoclonales anti IL17A, anti JAK1/2 y terapia fotodinámica.42
Pustulosis exantemática aguda generalizada
La pustulosis exantemática generalizada aguda es una reacción adversa cutánea grave a medicamentos, caracterizada por fiebre y rápida formación de pústulas estériles no foliculares sobre una base eritematosa. El periodo desde la exposición al fármaco hasta la aparición de la reacción suele ser de 48 horas (mediana de 24 horas).43
Fisiopatología
La pustulosis exantemática generalizada aguda es una enfermedad mediada por células T, clasificada dentro de las reacciones de hipersensibilidad de Gell y Coombs de tipo IVd.
Cuadro clínico
Se caracteriza por rápida aparición de cientos de pústulas pequeñas, estériles y no foliculares sobre una base eritematosa, pruriginosas, principalmente distribuidas en el tronco y las regiones intertriginosas. No suele afectar las mucosas, pero cuando sucede se limita a un solo sitio, por ejemplo, los labios o la mucosa bucal. La leucocitosis y la fiebre son características. Durante el proceso de curación existe descamación en la zona afectada.
Los fármacos asociados con mayor frecuencia se enumeran en el Cuadro 1.
Diagnóstico
El diagnóstico de pustulosis exantemática generalizada aguda se establece con base en criterios clínicos e histopatológicos. El grupo EuroSCAR desarrolló una puntuación de validación de la pustulosis exantemática generalizada aguda. Se trata de un esquema estandarizado basado en la morfología, curso clínico e histología que clasifica a los pacientes con sospecha de pustulosis exantemática generalizada aguda, y pustulosis exantemática generalizada aguda definida, probable, posible o sin la enfermedad (Cuadro 2). Puede llevarse a cabo la prueba de parche farmacológico para identificar la causa de la enfermedad cuando el medicamento responsable no es claro.44
Los hallazgos histológicos incluyen: pústulas intracorneales, subcorneales y/o intraepidérmicas, con edema dérmico papilar que contiene infiltrado de neutrófilos y eosinófilos.45
Cuadro 2 Score de validación de pustulosis exantemática generalizada aguda del estudio EuroSCAR
| Morfología | Puntos |
|---|---|
| Pústulas: | |
| Típicas* | + 2 |
| Compatibles** | + 1 |
| Suficientes*** | 0 |
| Eritema: | |
| Típico | + 2 |
| Compatible | + 1 |
| Insuficiente | 0 |
| Distribución: | |
| Típica | + 2 |
| Compatible | + 1 |
| Insuficiente | 0 |
| Curso: | |
| Afectación de mucosas: | |
| Sí | - 2 |
| No | 0 |
| Inicio agudo (<10 días): | |
| Sí | 0 |
| No | - 2 |
| Curación (<15 días): | |
| Sí | 0 |
| No | - 4 |
| Fiebre mayor de 38°: | |
| Sí | + 1 |
| No | 0 |
| Neutrófilos 7,000 /mm3 | |
| Sí | + 1 |
| No | 0 |
*Interpretación: 0 = no es PEGA, 1-4 = posible, 5-7 = probable, 8-12 = definido. NED = no especificado de otra manera. *Típico: morfología típica. **Compatible: morfología atípica, pero no sugestiva de otra enfermedad. ***Insuficiente: no es posible juzgar las lesiones (principalmente por el estado tardío de la enfermedad o mala calidad de las imágenes).
Tratamiento
El tratamiento se enfoca en suspender el fármaco causante (lo que alivia los síntomas en pocos días), y controlar la afectación sistémica y prevenir infecciones con apósitos húmedos y antisépticos durante la fase pustulosa. Debe evitarse la prescripción de antibióticos, a menos que haya infección de las pústulas. En pacientes resistentes al tratamiento se ha indicado ciclosporina, infliximab o secukinumab.46,47
Pronóstico
La muerte suele ser consecuencia de disfunción orgánica múltiple, y puede ocurrir en un 20% de los casos. Afecta el hígado, los riñones y pulmones, y existe coagulación intravascular diseminada; el riesgo de muerte es más alto en personas con comorbilidades y afectación extensa de las mucosas.48
Síndrome de reacción a medicamentos con eosinofilia y síntomas sistémicos (DRESS)
También conocido como síndrome DRESS, es una reacción adversa a medicamentos tipo IVb, según la clasificación de Gell y Coombs. Se trata de una reacción de hipersensibilidad tardía mediada por linfocitos T. La exposición al fármaco inicia el proceso de activación de células presentadoras de antígeno, que estimulan a los linfocitos T CD4, lo que genera la liberación de interleucinas y, a su vez, recluta y activa los eosinófilos (células clave en el DRESS).49
Epidemiología
Se estima que 1 de cada 1000 y 1 de cada 10,000 personas expuestas a ciertos fármacos padecerán síndrome DRESS, con mayor incidencia en las mujeres y la población afroamericana.50 Se han identificado alelos específicos de antígeno leucocitario humano relacionados con ciertos tipos de hipersensibilidad a fármacos. El alelo HLA-B 58:01 se ha relacionado con reacciones alérgicas al alopurinol, el HLA-B 15:02 con carbamazepina y el HLA-B 57:01 con abacavir.51,52
Cuadro clínico
Las manifestaciones clínicas y los hallazgos de laboratorio identificados en pacientes con síndrome DRESS aparecen de dos a ocho semanas de la administración continua del fármaco, inicialmente con fiebre, seguido de un exantema maculopapular característico. El síndrome se caracteriza por fiebre (100%), eosinofilia (95%), exantema (87%), linfocitosis atípica (67%) y linfadenopatía (54%). Los órganos más afectados suelen ser el hígado (94%) y los riñones (40%).53
Los fármacos más frecuentes relacionados se exponen en el Cuadro 1 54
Diagnóstico
El diagnóstico de síndrome DRESS se basa en indicadores clínicos, el antecedente de exposición a fármacos 2 a 8 semanas antes y los hallazgos de laboratorio. Para estandarizar el diagnóstico se aplican los criterios del RegiSCAR (Registro de Reacciones Adversas Cutáneas Severas a Medicamentos), que incluyen manifestaciones clínicas y resultados de laboratorio, y se considera un caso confirmado si la puntuación es igual o mayor de 6, probable si está entre 4 y 5, posible si se encuentra entre 2 y 3 puntos, y excluido si la puntuación es igual o menor de 2.55 Cuadro 3
Los hallazgos de laboratorio informan: linfocitos atípicos y eosinofilia mayor de 1500 cel/mm3 o más del 20%, incluso se consideran concentraciones significativas de 700 a 1499 cel/mm3 o del 10-19%. La biopsia evidencia dermatitis, queratinocitos apoptóticos y/o coxistencia de eosinófilos.56
Cuadro 3 RegiSCAR: sistema de puntuación para el diagnóstico de síndrome DRESS
| Puntaje | |||
|---|---|---|---|
| Manifestaciones | No | Sí | Desconocido |
| Fiebre mayor de 38.5° | -1 | 0 | -1 |
| Ganglios linfáticos (más de 2 sitios, mayor de 1 cm) | 0 | 1 | 0 |
| Linfocitos atípicos | 0 | 1 | 0 |
| Eosinofilia | |||
| 700 – 1,499 cel/mm3 o 10-19.9% | 0 | 1 | |
| >1,500 cel/mm3 o >20% | 2 | ||
| Erupción cutánea | |||
| Extensión mayor del 50% | 0 | 1 | |
| Al menos 2: edema, infiltración, púrpura, descamación | -1 | 1 | |
| Biopsia sugerente de síndrome DRESS | -1 | 0 | |
| Afectación de órganos internos | |||
| Uno | 0 | 1 | 0 |
| Dos o más | 2 | ||
| Resolución en >15 días | -1 | 0 | -1 |
| Evaluación de otras posibles causas* | 0 | 0 | |
| Puntaje total: | |||
| >6: Confirmado | |||
| 4 a 5: Probable | |||
| 2 a 3: Posible | |||
| <2: Excluido | |||
*DRESS: síndrome de reacción a medicamentos con eosinofilia y síntomas sistémicos. Adaptado de Sasidharanpillai S, Ajithkumar K, Jishna P, et al. RegiSCAR DRESS (drug reaction with eosinophilia and systemic symptoms) validation scoring system and Japanese consensus group criteria for atypical drug-induced hypersensitivity syndrome (DiHS): A comparative analysis. Indian Dermatol Online J. 2022;13(1):40. doi:10.4103/idoj.idoj_196_21.55
Tratamiento
En primera instancia deberá suspenderse el medicamento causante y comenzar con corticoesteroides sistémicos para tratar la fase aguda del síndrome DRESS, con lo que se ha observado alivio de la fiebre y el sarpullido de manera rápida.3
En casos graves, que no responden adecuadamente con los corticoesteroides, puede indicarse el tratamiento de segunda línea: ciclosporina, inmunoglobulina por vía intravenosa o tofacitinib inhibidor de quinasa Janus (JAK), especialmente en casos resistentes al tratamiento.57
La desensibilización a los fármacos está contraindicada en pacientes con síndrome DRESS, porque al ofrecer el medicamento causante puede empeorar la reacción.
Pronóstico
La mortalidad en pacientes con síndrome DRESS es del 20%, pero puede variar según la gravedad y el tratamiento oportuno. Se ha observado que la afectación hepática y renal aumentan el riesgo de mortalidad.58 El síndrome DRESS es una alteración seria que requiere una atención médica inmediata y tratamiento adecuado.
Síndrome de Stevens-Johnson y necrólisis epidérmica tóxica
Son alteraciones poco frecuentes, caracterizadas por necrosis epidérmica generalizada y desprendimiento de la piel. Suponen una urgencia dermatológica, con mecanismo fisiopatológico común. La clasificación de la enfermedad se establece con base en el porcentaje de superficie corporal afectada.Cuadro 4
Cuadro 4. Clasificación del síndrome de Stevens-Johnson/necrolisis epidérmica tóxica (SJS/NET) según porcentaje de superficie corporal afectada
| Enfermedad | Porcentaje de superficie corporal afectada |
|---|---|
| SJS | < 10% |
| Superposición del SJS/NET | 10–30% |
| NET | > 30% |
*Adaptado de Frantz R, Huang S, Are A, Motaparthi K. Stevens–Johnson Syndrome and Toxic Epidermal Necrolysis: A Review of Diagnosis and Management. Medicina (B Aires). 2021;57(9):895. doi:10.3390/medicina57090895.60
Fisiopatología
Este tipo de reacciones de hipersensibilidad se encuentran mediadas por linfocitos T (tipo IVc). La muerte de queratinocitos implica la interacción del ligando de Fas soluble (sFasL) y el receptor Fas en su superficie. La Granulisina, junto con la IL-15, tienen participación importante en la apoptosis, y las ampollas se relacionan con la gravedad de la enfermedad, aunque no son específicas. La Galectina-7 sérica y la quinasa -3 (RIP3) son biomarcadores específicos.59
Epidemiología
La incidencia varía de acuerdo con la situación geográfica. Es más frecuente en mujeres 1.5:1.59,60 Se ha demostrado que HLA-B57:01 tiene un valor predictivo negativo del 100% y valor predictivo positivo de 55% relacionado con el consumo de abacavir y la posibilidad de padecer síndrome de Stevens-Johnson y necrólisis epidérmica tóxica. Se ha estimado un 3% de valor predictivo positivo en caso de la ingesta de alopurinol.61-63
Cuadro clínico
Existe una etapa prodrómica con fiebre, malestar general, odinofagia y tos, seguida de afectación en dos o más mucosas y la piel, con máculas eritematosas o lesiones en diana atípicas en el tronco que progresan y confluyen con un centro oscuro, ampollas flácidas y signo de Nikolsky positivo con denudación cutánea.59,60 Se ha demostrado que HLA-B57:01 tiene un valor predictivo negativo del 100% y valor predictivo positivo de 55% relacionado con el consumo de abacavir y la posibilidad de padecer síndrome de Stevens-Johnson y necrólisis epidérmica tóxica. Se ha estimado un 3% de valor predictivo positivo en caso de la ingesta de alopurinol.60,64
Los fármacos asociados comúnmente con síndrome de Stevens-Johnson y necrólisis epidérmica tóxica se muestran en el Cuadro 1.65
Diagnóstico
Es importante elaborar la historia clínica completa, con descripción detallada del inicio de los síntomas, que suele aparecer entre los 4 días y 4 semanas de iniciar el medicamento. El algoritmo de ALDEN y de Liverpool se utilizan para identificar los fármacos causales. Debe evitarse la reexposición al medicamento causal.59,60 Se ha demostrado que HLA-B57:01 tiene un valor predictivo negativo del 100% y valor predictivo positivo de 55% relacionado con el consumo de abacavir y la posibilidad de padecer síndrome de Stevens-Johnson y necrólisis epidérmica tóxica. Se ha estimado un 3% de valor predictivo positivo en caso de la ingesta de alopurinol.60,66 La biopsia reporta licuefacción de la capa basal temprana, con queratinocitos necróticos y linfocitos. En etapas avanzadas existe separación subepidérmica, con necrosis epidérmica total.
Tratamiento
El tratamiento de primera línea consiste en incluye apoyo, administración de líquidos y electrolitos, suspensión del medicamento causante y tratamiento de las heridas de forma estéril. Se han indicado diferentes fármacos: corticosteroides, inmunoglobulina por vía intravenosa, ciclosporina, incluso infliximab y etanercept, pero sus resultados aún se discuten. Es importante evitar la reexposición al medicamento y llevar un brazalete con información de los medicamentos que causan las reacciones y con estructura química similar.60,65
Pronóstico
Las tasas de mortalidad varían del 4.8 al 9% para el síndrome de Stevens-Johnson y del 19.4 al 29% para la superposición del síndrome de Stevens-Johnson y necrólisis epidérmica tóxica, y del 14.8 al 48% para necrólisis epidérmica tóxica.59,60
Las escalas de gravedad en pacientes con síndrome de Stevens-Johnson y necrólisis epidérmica tóxica, por ejemplo: SCORTEN y ABCD-10 (como estala auxiliar), ayudan a establecer el pronóstico y orientan la atención en el área correspondiente, en piso o terapia intensiva de quemados. Cuadro 5
Cuadro 5 Escalas de gravedad en SJS/ NET de SCORTEN y ABCD-10
| SCORTEN | ABCD-10 | ||
|---|---|---|---|
| Parámetro | Valor | Parámetro | Valor |
| Edad > o = 40 años | 1 | Edad > o = a 50 años | 1 |
| Malignidad sí | 1 | Bicarbonato sérico < 20 mmol/L | 1 |
| % >10 de SC desprendido | 1 | Cáncer activo (sí) | 2 |
| Bicarbonato sérico < 20 mmol/L | 1 | Diálisis antes de la admisión (sí) | 2 |
| Nitrógeno ureico sérico > 28 mg/dL | 1 | % >10 de superficie corporal desprendida | 1 |
| Glucosa sérica > 252 mg/dL | 1 | ||
| Taquicardia > o = 120 lpm | 1 | ||
| Valor máximo | 7 | 8 | |
*El valor de Scorten de 0 a 1 traduce una mortalidad estimada de 31; la puntuación de 4 se correlaciona con 58.3% de mortalidad y la puntuación >5 con mortalidad mayor del 90%. Adaptada de las Referencias60,63,64
Enfermedad del suero-reacción de Arthus
Este tipo de reacciones inmunológicas se distinguen por erupción súbita morbiliforme, a menudo acompañadas de fiebre, luego de la exposición a fármacos proteicos o derivados séricos.
Fisiopatología
Las reacciones asociadas con esta enfermedad corresponden a hipersensibilidades tipo III de Gell y Coombs. Implican el depósito de complejos inmunes en los tejidos y vasos sanguíneos, con activación de la vía clásica del complemento, lo que genera la liberación de mediadores químicos que atraen neutrófilos, causando daño tisular, microtrombos y respuesta inflamatoria sistémica.67,68
Epidemiología
La incidencia real se desconoce, pero se estima en alrededor del 7% de las reacciones adversas a vacunas y medicamentos de este tipo, y aumenta con exposiciones repetidas.67 Suele afectar con mayor frecuencia a los pacientes pediátricos que a los adultos.68
Cuadro clínico
Este tipo de reacciones se expresa de forma tardía, entre 1 a 6 semanas posteriores a la administración del fármaco y no requiere sensibilización primaria. Pueden aparecer de forma localizada (reacción de Arthus) o con afectación sistémica (enfermedad del suero) y se caracterizan por erupción morbiliforme aislada, o asociada con urticaria; inicia con una banda eritematosa, progresiva hasta purpúrica que puede acompañarse de síntomas sistémicos: fiebre, artralgias y linfadenopatias localizada. Las manifestaciones menos frecuentes son mialgias, náuseas, vómito, cefalea, artritis, nefritis, vasculitis y, en casos graves, glomerulonefritis, miocarditis o coroiditis.67-70
También se han reportado casos con leucopenia o leucocitosis leve, elevación de la velocidad de sedimentación globular y concentraciones altas de complemento C3a.
Los fármacos más frecuentes relacionados aparecen en el Cuadro 1.70,71
Diagnóstico
El diagnóstico se establece con los siguientes criterios: tiempo de aparición de los síntomas, manifestaciones clínicas y ausencia de otras causas inmunológicas o infecciosas. Las pruebas de laboratorio reportan: leucopenia o leve leucocitosis, elevación de la velocidad de sedimentación globular y concentraciones bajas de complemento. Hasta el momento no existen estudios estandarizados para la confirmación diagnostica.68
Vasculitis inducida por medicamentos
Las vasculitis inducidas por medicamentos son un grupo de enfermedades vasculares de carácter inflamatorio, en las que se identifica un fármaco específico como causa de la alteración. Pueden limitarse a la piel o afectar órganos internos. El daño tisular por la inflamación vascular se clasifica según el tipo dominante de vasos implicados, es decir, de pequeño, mediano o grandes vasos.72
Fisiopatología
Representa un mecanismo de hipersensibilidad tipo III, con depósito de inmunocomplejos circulantes en la pared vascular, activación del complemento y posterior quimiotaxis de neutrófilos, que son los responsables del daño tisular.72,73
Epidemiología
No existen datos epidemiológicos acerca de la incidencia de las vasculitis inducidas por medicamentos, pero se estima un aproximado del 15 al 20% de las vasculitis cutáneas.74
Vasculitis de pequeños vasos
Puede resultar en vasculitis cutánea aislada o asociada con la afectación de otros órganos (riñón y nervios periféricos).72
a) Vasculitis leucocitoclástica cutánea inducida por medicamentos: Es la forma más común de la enfermedad.
b) Vasculitis por IgA inducida por medicamentos: es una alteración excepcional, con manifestaciones cutáneas, glomerulonefritis y sangrado gastrointestinal en algunos casos.
c) Vasculitis asociada con anticuerpos anticitoplasma de neutrófilo (ANCA) inducida por medicamentos: los pacientes con este tipo de alteración pueden manifestar solo vasculitis cutánea, o acompañarse de la afectación de un órgano principal, por ejemplo: enfermedad pulmonar intersticial, glomerulonefritis crecéntica semilunar, hemorragia alveolar difusa grave o vasculitis retiniana.75,76
Cuadro clínico
La vasculitis cutánea se manifiesta con urticaria, eritema, petequias, púrpura, pápulas purpúricas, vesículas y bullas hemorrágicas, nódulos, livedo racemosa, úlceras profundas en sacabocados y gangrena digital. Las lesiones cutáneas se correlacionan estrechamente con el tamaño del vaso afectado por la vasculitis.77
El Cuadro 1 muestra los fármacos más frecuentemente relacionados con las vasculitis inducidas por medicamentos.
Diagnóstico
El diagnóstico se estable con la obtención de una biopsia de piel, además de la historia clínica detalla y prescripción de medicamentos. El diagnóstico es de exclusión, de protocolo de vasculitis.77
Tratamiento
El tratamiento depende de la gravedad de la enfermedad. En pacientes con síntomas leves deberá suspenderse el fármaco implicado con la reacción, pues se ha demostrado que luego de establecer el diagnóstico puede haber curación de la enfermedad. Los pacientes con condiciones graves pueden recibir prednisona, incluso inmunosupresores (ciclofosfamida) o plasmaféresis.72,74,77
Citopenias autoinmunes
Los cambios en el recuento sanguíneo pueden ocurrir como reacciones adversas a medicamentos y engloban tres síndromes:
Anemia hemolítica por fármacos
Trombocitopenia inmune por fármacos
Agranulocitosis inducida por fármacos
Fisiopatología
La hemólisis y trombocitopenia son reacciones de hipersensibilidad tipo II, los anticuerpos dependientes o independientes del fármaco se enlazan al principio activo mediante células sanguíneas y provocan su destrucción.4,78 La agranulocitosis por fármacos es de tipo IV, con activación de células T dependientes de HLA. 79
Epidemiología
Son reacciones raras, con incidencia de 10 casos por cada 1,000,000 habitantes al año para trombocitopenia inmune por fármacos, de 7.2 casos por cada 1,000,000 habitantes para agranulocitosis inducida por fármacos, y de 1 caso por cada 1,000,000 habitantes al año para anemia hemolítica inducida por fármacos. El HLA-DQB1 y HLA-B158T se han asociado con agranulocitosis inducida por fármacos, debido al consumo de clozapina.79
Cuadro clínico
Las manifestaciones clínicas aparecen días o semanas después de la exposición al fármaco. Los pacientes con anemia padecen fatiga, disnea e ictericia.80 En quienes manifiestan agranulocitosis inducida por fármacos ocurre infección por patógenos poco comunes y puede evolucionar a choque.81 En sujetos con trombocitopenia provocada por fármacos puede haber púrpura, petequias y, en ocasiones, hemorragias digestivas, genitales, urinarias o intracraneales, con valores plaquetarios muy bajos. La trombocitopenia por heparina supone un alto riesgo de trombosis.
Los estudios de laboratorio informan hemólisis con anemia, reticulocitosis, hiperbilirrubinemia indirecta, DHL elevada con Coombs positivo. La agranulocitosis se caracteriza por concentraciones menores de 500 neutrófilos/μL y la trombocitopenia con plaquetas menores de 150,000/mm3, el sangrado aparece con recuentos menores de 50,000/mm3.4,81-87
Los fármacos más frecuentes relacionados con esta alteración se enlistan en el Cuadro 1.80-90
Diagnóstico
Las pruebas para detectar anticuerpos unidos a plaquetas o eritrocitos en presencia del fármaco no están estandarizadas, tienen buena especificidad, pero poca sensibilidad, y el resultado negativo no descarta el diagnóstico de la enfermedad.78 El diagnóstico se establece por las manifestaciones clínicas, y la detección de anticuerpos no debe retrasar el tratamiento.
Tratamiento
La interrupción del fármaco es la piedra angular del tratamiento. La mejora hematológica ocurre en 1 a 2 semanas. Los pacientes con anemia hemolítica inducida por fármacos grave requieren transfusiones; se han indicado corticoesteroides e inmunoglobulina por vía intravenosa, sin evidencia sólida.80 Los sujetos con agranulocitosis inducida por fármacos deberán aislarse y recibir antibióticos de amplio espectro; algunos pacientes pueden requerir filgrastim.79 Quienes padecen trombocitopenia inmune inducida por fármacos pueden requerir transfusión de concentrados plaquetarios, y quienes sufren trombocitopenia inmune inducida por heparina deberán recibir tratamiento con anticoagulantes.81,82,88-90
Pronóstico
La mortalidad en pacientes con anemia hemolítica inducida por cefalosporinas y diclofenaco se estima en 6 al 15%. En quienes padecen agranulocitosis inducida por fármacos es del 5%, concomitante con factores de riesgo como: edad mayor de 65 años, recuento de neutrófilos menores de 100 células/μL y signos de sepsis. Y la mortalidad en sujetos con trombocitopenia inmune inducida por fármacos varía del 1 al 5%, incluso es más alta en adultos.4
Conclusiones
El conocimiento sólido acerca de las reacciones adversas a medicamentos mejora el diagnóstico y reconocimiento de este tipo de alteraciones, además de aumentar la calidad de la atención al paciente. Incluso el conocimiento recopilado permitirá desarrollar tratamientos más seguros y efectivos en el campo médico. La obtención y el análisis de datos de las reacciones permiten la identificación de patrones, tendencias y factores de riesgo subyacentes, llevando a la comunidad científica a la comprensión profunda y actualizada de estas manifestaciones. La colaboración entre los profesionales de la salud e investigadores servirá de piedra angular para brindar una atención médica de calidad y sentará las bases de la investigación futura.

