











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
Hypokalemic periodic paralysis (HPP) is a rare genetic neuromuscular disorder inherited in an autosomal dominant pattern with incomplete penetrance1-7, predominantly affecting males with a ratio of 20:18, and an estimated incidence of 1:100,0009. It is known as Westphals disease2, and is more prevalent in Western countries8. The clinical presentation is variable, characterized by mild fatigue, paroxysmal episodes of transient or recurrent paresis, focal or generalized skeletal muscle flaccid paralysis, myalgias, or painless muscle weakness1,7-9. HPP is associated with low potassium levels (< 3.5 mmoL/L)1,4,10,11, due to a defect in the muscle ion channels1,3. While it can be life-threatening, it is completely reversible if diagnosed and treated appropriately in a timely manner2,8.
Weakness predominantly affects the lower extremities rather than the upper limbs, with proximal muscles being more frequently involved than distal ones, while facial muscles typically remain preserved7. Symptoms generally appear in early adolescence6, and impaired muscle function may be accompanied by respiratory dysfunction, electrocardiographic changes (QT prolongation and a flat T wave), cardiac arrhythmias7, rhabdomyolysis12, constipation, or sensory deficits7. Other forms of periodic paralysis include thyrotoxic periodic paralysis, hyperkalemic periodic paralysis, and Anderson syndrome2.
Although HPP is the most common type of periodic paralysis, it is rare, with a prevalence of 1 in 100,0001,5,7. In cases where there is no family history, diagnosis can be delayed and challenging. HPP demonstrates phenotypic variability regarding onset, duration, and presentation of symptoms, manifesting as transient episodes that fluctuate in duration and severity; however, most individuals achieve full recovery1.
HPP is caused by heterozygous pathogenic variants in the genes for the L-type voltage-gated calcium channel α1 S subunit (CACNA1S, OMIM *114208) located at 1q31-32 and the voltage-gated sodium channel α4 subunit gene (SCN4A, OMIM *603967) at 17q23-251,5,7,11, present in 70-80% and 10% of cases, respectively9.
Paralytic episodes may occur spontaneously during the second decade of life5,9, and can be secondary to conditions such as hyperthyroidism (Graves disease)1,3, thyrotoxicosis1,7, distal renal tubular acidosis1,3,7,13, Bartter's syndrome13, Gitelmans syndrome, primary hyperaldosteronism7,13, Cushing's syndrome13, diabetes insipidus, celiac disease1, Crohn disease14, and the use of drugs such as diuretics7, and cephalexin15. Triggering factors include resting state after intense exercise, high carbohydrate diets2,4,5,7,9,16, alcohol cosumption9, mental stress7, prolonged fasting2,7, acute febrile illness7, pregnancy, and menstruation4. Episodes often appear several hours after exposure to these aggravating factors, which increase the release of insulin or epinephrine, promoting the movement of potassium into cells and resulting in hypokalemia. For this reason, lifestyle changes are crucial to avoid exacerbations and prevent severe acute symptoms. It is also vital to consider the patients history and establish adequate differential diagnoses to identify and correct potential causes of symptoms promptly7.
We present a new family case of HPP, emphasizing the importance of understanding the etiological cause to guide available therapeutic options and provide genetic counseling for the family.
Clinical case
The proband was a 16-year-old male adolescent with a history of flaccid paralysis associated with severe physical exercise since the age of 12. He had been monitored and followed by pediatric neurology. He was the product of non-consanguineous parents, following a second pregnancy. There was a history of hypotonia in childhood that warranted physical therapy, with satisfactory improvement, leading to independent walking by 2 years of age. He maintained adequate academic performance and participated in water polo practice.
He was admitted after manifesting a rapidly progressive episode of flaccid paralysis affecting the trunk and all four extremities, with symmetrical, painless muscle weakness, and fatigue lasting 12 h, accompanied by preserved osteotendinous reflexes and a normal sensory examination. Examination of other systems revealed unremarkable findings, and laboratory tests indicated hypokalemia of 2.1 mEq/L (normal range 3.5-5.10), following intense physical activity and subsequent confirmation of COVID-19 infection. Serum chloride and sodium levels were within the normal limits. Other hematological and biochemical parameters, arterial blood gas analysis, and thyroid hormones were normal. He was treated with parenteral potassium chloride supplementation, and serum potassium levels normalized after 24 h. Within 2 days, his weakness completely resolved, and he was monitored under pediatric observation without complications during his inpatient treatment. The electrocardiogram upon admission showed QT prolongation, shallow T waves, and prominent U waves consistent with hypokalemia, whereas a repeat electrocardiogram normalized after treatment. The patient was discharged with oral potassium supplements.
Family history revealed that the maternal grandfather had a similar history of flaccid paralysis affecting all limbs, associated with muscle weakness and abnormal breathing with diminished tendon reflexes lasting around 6 h, along with hypokalemia at 5, 38, and 40, with seven episodes occurring within a month during the latter period. In addition, the maternal half-brother, under 12 years old, experienced a first episode of paralysis following intense physical activity with hypokalemia of 2.5 mmoL/L. A second episode occurred 10 months later during a period of psychological stress. The mother reported experiencing anesthetic complications following maxillofacial surgery at age 18, an event that also affected her sister and father during anesthesia (Fig. 1).
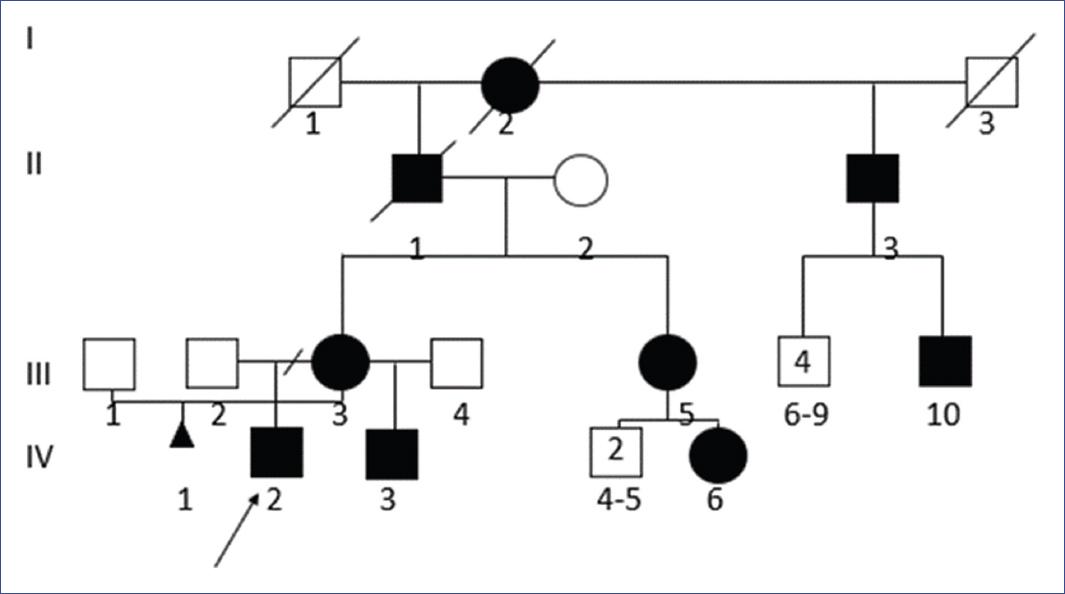
Figure 1 Genealogy of the studied family. The arrow points to the purpose (IV: 2), his mother (III: 3), and maternal half-brother (IV: 3) presented the CACNA1S gene variant c.2700G > A (p.Arg900Ser). An autosomal dominant inheritance pattern is demonstrated in the presence of male-male transmission (II:3 and III:10
Considering the personal and family medical history, genetic testing for genes associated with HPP was performed on the patient through a Next Generation Sequencing panel, identifying a heterozygous pathogenic variation in the CACNA1S gene (c.2700G>A; p.Arg900Ser). The proband's mother and her younger maternal half-brother also exhibited this variant. In addition, a likely pathogenic variant in the donor splice site of the TNNT1 gene (c.387 + 1G>A) was identified in the patient. Genetics follow-up has been conducted to provide family genetic counseling.
Methods
Genomic DNA obtained from the submitted sample was enriched for targeted regions using a hybridization-based protocol and sequenced using Illumina technology. All targeted regions were sequenced with a depth of ≥ 50x or supplemented with additional analysis. Reads were aligned to a reference sequence (GRCh37), and sequence changes were identified and interpreted in the context of a single clinically relevant transcript. Enrichment and analysis focused on the coding sequences of the indicated transcripts, along with 10bp of flanking intronic sequence and other specific genomic regions known to be causative of disease at the time of assay design. Exonic deletions and duplications were identified using an in-house algorithm that compares the read depth for each target in the proband's sequence with both the mean read depth and distribution from clinical samples. Markers across the X and Y chromosomes underwent analysis for quality control purposes, detecting deviations from the expected sex chromosome complement, which may be reported in accordance with internal guidelines. All clinically significant observations were confirmed using orthogonal technologies, except individually validated variants and those previously confirmed in a 1st-relative. Confirmation technologies include Sanger sequencing, Pacific Biosciences single-molecule real-time sequencing, MLPA, MLPA-seq, and Array CGH. Array CGH confirmation of Next Generation Sequencing CNV calling performed by Invitae Corporation. If a CNV is identified, MLPA or MLPA-seq was conducted to confirm the variant.
An rsID is a unique identifier that refers to a single genomic position and is used to associate population frequency information with sequence changes at that position. Reported population frequencies are derived from various public sites aggregating data from large-scale population sequencing projects, including ExAC, gnomAD, and dbSNP.
A MedGen ID is a unique identifier for articles in MedGen, NCBI's centralized database for information on genetic disorders and phenotypes. Search using the MedGen ID at the NCBI site.
The genes of the selected panel were 52: ACTA1, ANO5, ATP2A1, BAG3, BIN1, CACNA1S, CAV3, CCDC78, CFL2, CNTN1, COL12A1, COL6A1, COL6A2, COL6A3, CPT2, CRYAB, DES, DNAJB6, DNM2, DYSF, FHL1, FKBP14, FLNC, GNE, GYS1, KBTBD13, KCNJ2, KLHL40, KLHL41, LDB3, LMNA, LMOD3, MATR3, MEGF10, MTM1, MYH7, MYL2, MYOT, MYPN, NEB, RYR1, SCN4A, SELENON, SQSTM1, STAC3, STIM1, TIA1, TNNT1, TPM2, TPM3, TTN, and VCP.
Discussion
HPP type 1 (OMIM #170400) and type 2 (OMIM #613345) are rare inherited disorders of skeletal muscle channelopathy caused by dysregulation of sarcolemma excitability17, belonging to a group of genes that code for ion channels18. Most mutations involve substitutions of a positively charged arginine in the fourth transmembrane segment within the voltage sensor domains of calcium and sodium channels, respectively17.
Patients with HPP may experience persistent muscle weakness during episodes when muscle fibers become non-excitable due to defects in the potassium flux1. If hypokalemia is accurately diagnosed and corrected, symptoms can be suppressed, and reversible recovery can be achieved. However, if the diagnosis is delayed or incorrect, the patient may experience arrhythmias or cardiac arrest7. HPP can also present with motor deficits and respiratory failure, negatively impacting quality of life1. A family history can facilitate diagnosis.
The treatment strategy begins with patient education and lifestyle modifications to avoid triggering stimuli, minimizing the frequency and severity of attacks. Small, frequent meals with a low-sodium and carbohydrate diet, coupled with avoidance of strenuous exercise, can help reduce episodes. Chronic treatment with carbonic anhydrase inhibitors, notably acetazolamide, has been used empirically for over five decades1,4. However, it has been ineffective in some cases and occasionally worsens symptoms7. Dichlorphenamide could be considered for patients unresponsive to acetazolamide4. Treatment for acute symptoms includes plasma potassium replacement2,4, maintenance of acid-base balance, cardiac monitoring, and careful use of neuromuscular blocking agents2. Potassium-sparing diuretics such as spironolactone, triamterene1,4, eplerenone, or amiloride, along with some antiepileptic drugs such as topiramate, have emerged as effective alternatives for cases unresponsive to acetazolamide1. Attempting to manage paralysis solely through potassium supplementation can lead to rapid potassium release from cells, potentially resulting in rebound hyperkalemia.
Early diagnosis is essential for determining an effective treatment plan, as non-selective β-blockers can treat hypokalemia while simultaneously mitigating the risk of rebound hyperkalemia11. The long-term outlook for HPP patients is positive; however, they are sensitive to general anesthesia and may develop muscle paralysis and respiratory weakness. Proximal myopathy, malignant hyperthermia, and cardiac arrhythmias can pose life-threatening risks4.
Changes in electrolytes and acid-base status can aid in the early differential diagnosis of primary and secondary HPP. In the case of HPP secondary to conditions other than hyperthyroidism is more likely to lead to acid-base imbalance, presenting with higher pH and HCO3 levels, requiring larger doses of potassium supplementation, and taking longer for serum potassium levels to normalize and for patients to regain full muscle strength compared to those with primary or thyrotoxic HPP. Thus, arterial blood gas analysis may assist in the early differential diagnosis of primary and secondary HPP19.
SARS-CoV-2 infection can induce dysfunction in many organs through ACE2 receptor involvement and the consequences of the cytokine storm; this relationship is relevant in thyroid diseases. A documented case indicates hyperthyroidism with SARS-CoV-2 infection leading to refractory hypokalemia, managed with potassium replacement therapy over 2 months. Although the patient lacked a history of thyroid disease, laboratory results indicates hyperthyroidism. Following oral thiamazole, the thyroid hormone parameters improved, potassium levels normalized, and limb weakness stopped20. This represents the fourth documented HPP case associated with SARS-CoV-2 infection without thyroid abnormality, suggesting that this infection can precipitate HPP20-22.
The presence of a family history helps facilitate diagnosis. The CACNA1S gene encodes the voltage-gated calcium channel α subunit, which plays a critical role in calcium-mediated excitation and contraction coupling6,12, activating RYR1 to induce calcium release from the sarcoplasmic reticulum. Dominant mutations in the CACNA1S gene are associated with HPP, malignant hyperthermia, and congenital myopathy12. Multiple nonsense variations have been identified in HPP patients6, presenting variable phenotypes12. Genetic variation typically causes arginine replacement with an uncharged residue in the S4 segment of the channel. Available data supporting the notion to gate pore current affirms the episodic loss of fiber excitability during weakness attacks prevalent in HPP17.
The variation found within the studied family replaces an arginine residue with an uncharged residue in the S3 segment of the channel; this genetic variation in the CACNA1S gene was previously described by Jia et al.23 in an Asian patient, who exhibited substantial muscular edema in the lower legs, particularly within the medial, lateral, and soleus gastrocnemius muscles during the post-exercise period. Similarly, the same CACNA1S gene variation was noted in a Chinese family whose patients did not respond to acetazolamide but improved with combined treatment with triamterene and potassium supplements, leading to a reduced frequency of muscle weakness attacks24. All male carriers of the p.Arg900Ser variation experienced attacks, though all three were asymptomatic.
The genetic variation identified in the TNNT1 gene was found solely in the proband and is associated with Amish-type nemaline myopathy 5, which typically follows an autosomal recessive inheritance pattern. The variant in the proband was in a heterozygous state, indicating a likely gene carrier; this variation is not responsible for the described phenotype.
Conclusions
The CACNA1S gene variation identified in this family has not been previously reported in Caucasians. The genetic basis of HPP can influence treatment responses to various medications, suggesting that precise genetic diagnosis allows for personalized treatment. In this case, both the proband and his maternal half-brother began potassium and triamterene supplementation with favorable clinical outcomes, consistent with earlier reports24. Consequently, it is crucial to document therapeutic successes associated with specific genotypes to facilitate personalized medical care and establish a future consensus on treatment options for HPP while offering family genetic counseling.

