











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
A nivel global, el cultivo de haba abarca actualmente alrededor de 2.5 millones de hectáreas con una producción de 5.4 millones de toneladas y un rendimiento promedio de semilla de 2.1 toneladas/ha (FAOSTAT, 2023). Este cultivo no solo es valioso por sus propiedades alimenticias y medicinales que ofrece beneficios significativos para la salud humana (Prabhu y Rajeswari, 2018), sino también por su contribución económica. Sin embargo, los virus fitopatógenos representan una amenaza considerable para su producción (Mansourpour et al., 2022); (Makkouk et al., 1994).
La adopción de la PCR (Polimerase Chain Reaction) y otras técnicas de detección viral revelaron la complejidad de las etiologías virales en plantas (Binnicker, 2015). La introducción de la secuenciación de nueva generación (NGS, NextGeneration Sequencing) ha revolucionado la investigación genómica empleando datos biológicos masivos permitiendo analizar vastas cantidades de secuencias de forma eficiente (Rubio et al., 2020). Esta tecnología ha facilitado el diagnóstico avanzado y la identificación de viromas, es decir, el conjunto de virus y viroides presentes en plantas infectadas o sus vectores, abriendo nuevas opciones para su manejo (Barba et al., 2014).
En este contexto, el presente estudio se enfocó en parcelas de haba en Montecillo, Texcoco, Estado de México, donde se observó una prevalencia total de virosis caracterizada por síntomas como mosaico, moteado, enrollamiento de hojas, marchitamiento y disminución del crecimiento. Estos síntomas se traducen en una reducción significativa del rendimiento y la calidad de la semilla. El objetivo de esta investigación fue determinar el viroma asociado a estos síntomas, con la intención de contribuir al desarrollo de estrategias de manejo más efectivas para este cultivo. Se realizó un muestreo al azar de plantas de haba de 95 días de edad con síntomas de moteado, mosaico, enrollamiento foliar y marchitez en un lote ubicado en Montecillo, Texcoco, Estado de México. Se recolectaron hojas del tercio superior de 10 plantas que se conservaron en refrigeración a 4 °C hasta el proceso de extracción de ARN. Para la extracción del ARN total se maceró 0.1 g de tejido foliar de cada planta con nitrógeno líquido hasta obtener un polvo fino; utilizando el kit SV Total RNA Isolation System® PROMEGA de acuerdo con las instrucciones del fabricante. La calidad y concentración del ARN total extraído se cuantificó por espectrofotometría (Nanodrop 2000®, Thermo Scientific™). Para verificar la integridad se realizó electroforesis a 90 V durante 30 minutos en un gel de agarose al 1 % teñido con bromuro de etidio visualizado en un fotodocumentador de luz ultravioleta.
Para validar los resultados obtenidos mediante secuenciación masiva, se diseñaron cebadores específicos enfocados en los segmentos genómicos que codifican la replicasa viral y la proteína de la cubierta (Cuadro 1. Lista de los cebadores empleados). La selección de estos segmentos como blancos para el diseño de los cebadores se debe a su importancia funcional y su conservación entre diferentes aislamientos del virus, lo que los convierte en regiones ideales para la detección precisa del fitopatógeno. Una vez diseñados los cebadores, se procedió a realizar la reacción en cadena de la polimerasa con transcripción inversa (RT-PCR) para cada una de las muestras individuales que originalmente se combinaron para formar la muestra compuesta analizada por secuenciación masiva, además del resto de muestras que permanecieron en resguardo para analizar la presencia o ausencia de las especies encontradas.
Cuadro 1 Cebadores diseñados para la corroboración de la presencia de Potyvirus phaseoluteum (Pp) y Orthotospovirus impatiensnecromaculae (Oi) en tejido foliar de haba con síntomas de mosaico, moteado, enrollamiento foliar, marchitamiento y disminución del crecimiento.
| Virus | Iniciadores | Región que amplifica | Amplicón |
|---|---|---|---|
| Oi | S: 5’CAGCCGCTTCTTCACTCTGT3’ As: 5’TGTGAGTTTCTCGGAAGGCT3’ | Replicasa viral | 438 pb |
| Pp | S: 5’AATGCGAAGCCAACATTCCG3’ As: 5’CACGGTTGACATCTCCTGCT3’ | Nucleocápside | 319 pb |
La mezcla para la síntesis de cADN de los virus encontrados consistió en 5.3 μL de agua libre de nucleasas, 0.5 μL de cada cebador y 1 μL de RNA total para tener un volumen final de 7.3 μL que se incubaron por 5 minutos a 70 °C; finalizado este paso los tubos se colocaron en hielo. Posteriormente, se preparó una mezcla de 2 μL de amortiguador RT-5X, 0.5 μL de dNTP Mix y 0.2 μL de M-MLV Reverse Transcriptase; se adicionaron 2.7 μL de esta mezcla a cada tubo de reacción incubado en el primer paso para tener un total de 10 μL. Los tubos permanecieron durante 60 minutos a 42 °C y 10 minutos a 72 °C en el termociclador. Para la reacción de PCR la mezcla consistió de 2 μL de amortiguador 5X Green GoTaq®, 0.6 μL de MgCl2 (25 mM), 0.2 μL de dNTP Mix (10 mM), 0.6 μL de cada cebador (10 pmol/μL),
4.9 μL de agua libre de nucleasas, 0.1 μL de GoTaq® Flexi Polimerase (5u/μL) y 1 μL de cDNA para tener un volumen total de 10 μL por reacción. Las condiciones de amplificación fueron: ciclo inicial de desnaturalización de 5 min a 95 °C, 35 ciclos de 30 s a 95 °C, 30 s a 55 °C y 30 s a 72 °C seguido de una extensión final de 10 min a 72 °C. El producto de PCR se sometió a electroforesis a 80 V durante 55 minutos en un gel de agarosa al 1 % teñido con bromuro de etidio y se visualizó en un fotodocumentador de luz ultravioleta. Los amplicones obtenidos se enviaron a Macrogen Inc. (Corea) para su secuenciación y posterior comparación con las depositadas en el GenBank con BLASTn.
Para cada uno de los virus encontrados (P. phaseoluteum y O. impatiensnecromaculae) se realizaron dos análisis, para el segmento que codifica la replicasa viral y el que codifica la nucleocápside. Los genomas completos de los virus obtenidos se alinearon mediante ClustalW en MEGA X. Este alineamiento incluyó secuencias de genomas completos correspondientes a virus de la misma especie, de una especie diferente pero dentro del mismo género, y de una especie de un género diferente. La inclusión de estas secuencias, todas disponibles en la base de datos del NCBI, proporciona un marco comparativo que facilita la interpretación de las relaciones evolutivas entre los virus estudiados. La construcción del árbol filogenético se basó en la aplicación de modelos evolutivos sofisticados, específicamente el General-Time-Reversible (GTR) y Jones Taylor Thornton Model (JTT). Estos modelos se seleccionaron por su capacidad para estimar de manera precisa las tasas de sustitución de bases a lo largo del tiempo evolutivo, ofreciendo una visión detallada de las relaciones filogenéticas. Ambos se construyeron por el método de Máxima Verosimilitud con 500 repeticiones de bootstrap en MEGA X. Este enfoque estadístico permite evaluar la robustez de cada nodo del árbol, proporcionando un indicador de la confianza de las agrupaciones filogenéticas observadas.
Las evaluaciones de calidad de las muestras de ARN revelaron que se encontra-
ban dentro de los rangos óptimos de pureza, con relaciones de absorbancia 260/280 entre 1.8 y 2.2, y 260/230 también dentro del intervalo ideal, lo que indica una mínima presencia de contaminantes orgánicos e inorgánicos. Además, la concentración del ARN extraído fue superior a 300 ng/μL, superando el umbral requerido para asegurar una cantidad suficiente para análisis subsiguientes. Asimismo, en el gel de electroforesis para integridad se observaron bandas nítidas sin arrastre por fragmentación de las cadenas, lo que indica una integridad óptima.
Se obtuvieron dos genomas completos de Potyvirus phaseoluteum, uno de 9,577 y otro de 9,575 nt correspondientes a aislamientos distintos, denotados como CP1 MEX y CP2 MEX, respectivamente; asimismo, se obtuvieron los tres segmentos completos del genoma de Orthotospovirus impatiensnecromaculae cuyas longitudes fueron las siguientes: segmento L (replicasa viral) 8,763 nt, segmento M (proteína de movimiento) 3,629 nt y segmento S (nucleocápside proteica y proteína NSs) 1,825 nt. En los tres casos, las regiones codificantes se encontraron completas y se depositaron en el GenBank con los números de acceso PP098739 (BYMV CP1MEX), PP098740 (BYMV CP2MEX), PP098741 (INSV CPMEX L), (PP098742) INSV CPMEX M y PP098743 (INSV CPMEX S).
Se obtuvieron los amplicones correspondientes al tamaño esperado para cada virus en muestras de campo (Figura 1) con los indiciadores diseñados para los dos aislamientos de P. phaseoluteum y O. impatiensnecromaculae encontrados en esta investigación (Cuadro 1).
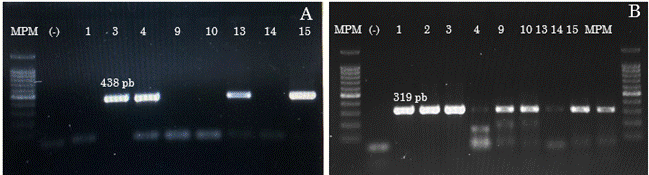
Figura 1 Detección de Orthotospovirus impatiensnecromaculae y Potyvirus phaseoluteum en plantas de haba mediante RT-PCR.
Producto de RT-PCR de un fragmento de 438 pb del segmento L que codifica la replicasa viral de O. impatiensnecromaculae: MPM= Marcador de peso molecular 100 pb PROMEGA, control negativo (-). Carriles 1, 3, 4, 9, 10, 13, 14 y 15 muestras de plantas de haba con síntomas de mosaico, moteado, enrollamiento y marchitamiento. B) Producto de RT-PCR de un fragmento de 319 pb del ORF del P. phaseoluteum que codifica para la nucleocápside: MPM= Marcador de peso molecular 100 pb PROMEGA, control negativo (-). Carriles 1, 2, 3, 4, 9, 10, 13, 14 y 15 plantas de haba con síntomas de mosaico, moteado, enrollamiento y marchitamiento. Las secuencias de los amplicones obtenidos tuvieron un 99% de cobertura y 99 % de identidad con aislados de P. phaseoluteum y O. impatiensnecromaculae depositados en el GenBank, respectivamente.
Los aislados CP1MEX y CP2MEX de BYMV encontrados en haba en esta investigación, se agrupan con aislados de Sudán e Irán, respectivamente, pero no entre ellos mismos (Figura 2).
El emparentamiento de los aislados de Potyvirus phaseoluteum con los de países asiáticos sugiere que el virus ha sido dispersado a otros continentes debido muy probablemente a la movilización de semilla, al ser ésta el principal medio de propagación del haba y constituir una forma de transmisión común de las especies del género Potyvirus (Díaz et al., 2010). De esta forma, una vez presente la fuente de inóculo en la plantación, su diseminación puede darse a través de diferentes especies de áfidos (Rodríguez-Pardina et al., 2019) lo cual explicaría la alta incidencia de plantas con síntomas observada en campo. No hay información sobre la presencia de P. phaseoluteum en México, por lo que se considera éste su primer reporte. Por otra parte, las secuencias de los segmentos L y S de INSV CPMEX obtenidas en este estudio (Figuras 3 y 4) no se agruparon con las reportadas en otros países, lo cual sugiere que se trata de un aislamiento altamente divergente, que puede atribuirse a su adaptación al espacio geográfico en que se encuentra derivado de los cambios que ha experimentado al paso del tiempo en la región de estudio.
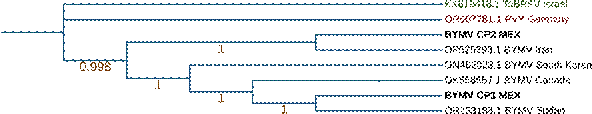
Figura 2 Árbol filogenético de los aislados de Potyvirus phaseoluteum CP1MEX y BYMV CP2MEX utilizando el modelo General-Time-Reversible y el método de máxima verosimilitud con 500 repeticiones de bootstrap.
impatiensnecromaculae infecta principalmente especies ornamentales y en México se ha reportado también en jitomate y chile (González-Pacheco y SilvaRosales, 2013). En un estudio previo se inocularon diferentes especies vegetales con O. impatiensnecromaculae y se encontró que el haba es un hospedante de este
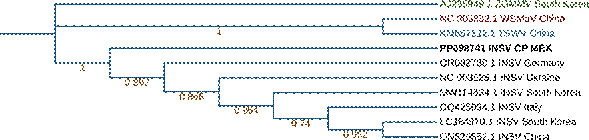
Figura 3 Árbol filogenético del segmento L correspondiente a la replicasa viral del aislado de Orthotospovirus impatiensnecromaculae INSV CP MEX utilizando el modelo Jones Taylor Thornton Model y el método de máxima verosimilitud con 500 repeticiones de bootstrap.
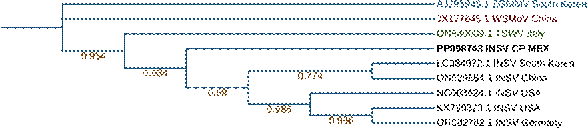
Figura 4 Árbol filogenético del segmento S correspondiente a la nucleocápside del aislado INSV CP MEX del Orthotospovirus impatiensnecronmaculae utilizando el modelo Jones Taylor Thornton Model y el método de máxima verosimilitud con 500 repeticiones de bootstrap.
virus (Abe El-Wahab y Elnagar, 2008). En contraste, en la presente investigación se reporta la infección natural de este virus en haba.
A nivel mundial se ha reportado a Fabavirus alphaviciae (antes broad bean wilt virus) (Balsak et al., 2023; Xia et al., 2020), Bromovirus BBMV (antes broad bean mottle virus) (Makkouk et al., 1988; Romero et al., 1993), Luteovirus phaseoli (antes bean leaf roll virus) (Makkouk et al., 2012 ), Potyvirus phaseoluteum (antes bean yellow mosaic virus) (Chatzivassiliou, 2021; El-Helaly et al., 2016) y Nanovirus necroflaviviciae (antes faba bean necrotic yellows virus) (Mansourpour et al., 2022; Katul et al., 1993) como especies principales que infectan naturalmente al haba, de las cuales sólo el P. phaseoluteum además del O. impatiensnecromaculae fueron encontrados en el presente estudio. Con base en estos resultados, sería necesario estudiar el viroma del haba en otras zonas productoras para conocer la diversidad de virus asociados a este cultivo en el país.
El estudio del viroma presente en cultivos de haba en Montecillo, Texcoco, Estado de México, ha revelado una composición específica de fitopatógenos virales, consistente en dos aislamientos distintos de Potyvirus phaseoluteum (designados como BYMV CP1MEX y BYMV CP2MEX) asociados a síntomas de mosaico y moteado y un aislamiento de Orthotospovirus impatiensnecromaculae (INSV CP MEX) asociado a enrollamiento foliar y marchitamiento en infección mixta con
O phaseoluteum. Este análisis marca la primera identificación documentada tanto del P. phaseoluteum como de la infección natural por O. impatiensnecromaculae en plantaciones de haba dentro del territorio mexicano. Estos hallazgos aportan evidencia crucial, subrayando la importancia de monitorear y gestionar estas infecciones virales para proteger la producción agrícola de haba en la región.

