











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
The identification of this family of malignant tumors was first described 100 years ago, when James Ewing first described a bone hemangioendothelioma, but it was not until 1973 that the term neuroectodermal tumor was coined by Hart and Earle, after the identification of a malignant tumor originating from the neuroectoderm.
The term -Tumors of the Ewing Sarcoma Family is currently used to define a group of tumors with a neuroectodermal origin that shares to a series of common characteristics, all of these primary malignant neoplasms constituted by the uniform proliferation of round blue cells of small size, with a round nucleus of fine chromatin, scarce cytoplasm, which do not produce extracellular matrix and which, as its name says, derives from the neural crest; they only differ from each another by the degree of neuroectodermal differentiation, which varies from poorly differentiated to obvious neuroectodermal differentiation with the presence of Homer-Wright roses (circle of grouped cells with a peripheral crown of nuclei) and by the presentation of the clinical picture1,2.
Tumors of the Ewing sarcoma family are the second most frequent cause of the malignant bone tumor, in the pediatric age; however, these tumors can occur at any age, with a peak incidence reported in children under 20 years, and with a slight male predominance3. However, this type of tumors located in soft tissues, present an incidence at later ages, and not present gender differences, nonetheless, they have a low incidence, representing 4% of the total of soft-tissue tumors4.
There is little information about the etiological factors of these neoplasms since a family association is discarded, relationship with a history of radiation can be ruled out, and there is no link with environmental factors either4,5. At present, within the oncogenesis of these types of tumors, the insulin-like growth factor receptor (IGF-IR) has been associated, which can be stimulated by ligands produced by tumor cells, resulting in the inhibition of apoptosis which entails irregularities in growth and transformation cell6. However, this area of knowledge about the tumor remains a subject of study.
Tumors of the Ewing family are neoplasms associated with the chromosomal translocation of the long arm of chromosomes 11 and 22 that result mainly in the fusion of FLI-1 and EWS genes, which can be observed in up to 85% of cases, the rest of the cases may be associated with a genetic fusion between EWS/ERG5.
The diagnosis by imaging studies is extremely complex, the computed tomography (CT) and the nuclear magnetic resonance imaging (MRI) allow us to have the visualization of the tumor panorama, knowing exactly the location, morphology, exact size, margins, and blood supply, however, imaging studies do not allow us to make a diagnosis because of the variability of the image that they can present and because of the similarities that the images can present with other types of malignant tumors5.
Conventionally, the histological diagnosis of this neoplasm was made by exclusion, but this situation has changed in the past years with the immunohistochemical markers. The markers include CD99, FLI-1, Caveolina-1 (CAV-1), and HNK-1 (CD57). When the four antibodies previously mentioned are grouped, practically, the diagnosis of more than 99% of cases is covered6,7.
The treatment for this group of tumors is usually combined because of the low response rate, it is performed with surgery, radiotherapy, and polychemotherapy. However, despite the treatment, the prognosis for this type of neoplasms is very poor, with disease-free survival at 5 years of 33%7.
Case report
This is a 47-year-old female patient, a resident of the city of Saltillo, Coahuila, Mexico, a teacher by profession. The patient has a hereditary family history associated with breast cancer and cervical cancer from two aunts. The rest of the antecedents without relevance for the case.
The current condition starts in May 2018, the patient with a pregnancy of 8 weeks of gestation for the date of the last menstrual period, which starts with the sudden onset of transvaginal bleeding with waste, thats why she goes to a private gynecology consultation. The doctor, after the clinical evaluation, diagnoses an incomplete abortion due to an embryonic pregnancy, so she goes to his social security health service.
The patient is diagnosed again by the gynecology service of the unit and determines the same diagnosis so they proceed to perform an instrumented uterine curettage. During the complete gynecological examination of the patient, mass is perceived in the left inguinal region, referred to as painless, of a month of evolution, with growth in the past days, so it is requested a TC and interconsultation to the general surgery service for the performance of a biopsy is requested.
The abdomen CT with double contrast revealed a tumor of heterogeneous appearance, with hypodense areas inside that indicate necrosis in the left inguinal region with suspicion of malignancy with measures of 69.3 × 52.12 mm. Therefore, an interconsultation with medical oncology and oncological surgery is requested.
It was performed a fine-needle aspiration biopsy of the lesion, reporting it extremely cellular, constituted of small hyperchromic basophilic cells with scarce cyanophilic cytoplasm, associated to changes in necrosis and with an inflammatory component, with a positive final diagnosis to neoplasia small round cells and blue color, compatible with endometrial sarcoma, a Mullerian tumor discarded.
During the medical oncology evaluation, laboratory studies are requested with results of tumor markers in normal parameters.
Surgical excision of the mass is performed by the surgical oncology service. The pathology report reveals macroscopic examination of an ovoid mass of 6 × 5 × 3 cm, solid, off-white color, with poorly defined edges and with areas of necrosis and hemorrhagic degeneration. The preliminary diagnosis of a primitive neuroectodermal tumor is given to the microscopic examination and immunohistochemical techniques are recommended for proper tumor classification.
The patient is referred to the medical oncology for radiotherapy sessions for local treatment in Mexico city since she is not a candidate for chemotherapy treatment due to the delay in the definitive histopathological report of the immunohistochemical studies.
Pathology samples are referred at the third level of attention for immunohistochemical studies, with positivity for CD99 and FLI-1, thus the diagnosis of a primitive neuroectodermal tumor is reinforced.
After the radiotherapy sessions, the patient go to a follow-up appointment with the oncological surgery service in June 2019, in which is detected the presence of a subdermal injury in the scalp in the right parietal area of 4 × 4 cm, referred with 15 days of evolution, the patient is referred asymptomatic, and without tumor activity data, so a skull CT is requested for the lesion study protocol.
The simple and contrasted skull CT reveals soft-tissue tumor located in the right parietal region with an intracranial component, with contrast enhancement, with measures of 39.7 × 46.6 mm, with lesions of lytic aspect in bone involved, with the possibility of being metastatic, therefore, MRI of the skull is requested to extend the assessment of the lesion.
In NMR, occupant lesion in the right parietal area is revealed, with intra- and extracranial component, heterogeneous lesion, hypodense concerning to the parenchyma (Figs. 1-3), whereby the interconsultation with the neurosurgery service is requested.

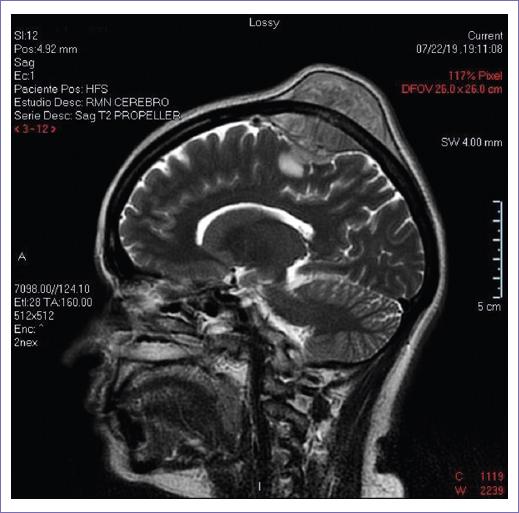

The patient begins to report headaches during the hospital stay, followed by hemiparesis of the left hemibody, with tremors and loss of strength, accompanied by ambulation difficulty.
The patient is surgically operated by the neurosurgery department, making an incomplete partial excision of the lesion in the right parietal area (Figs. 4 and 5).

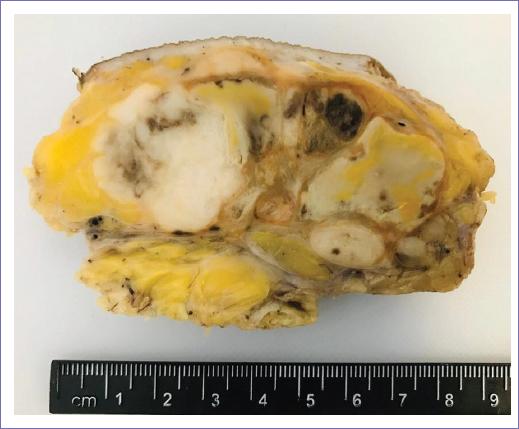
The extracted piece for the histopathological analysis is sent, which reveals the same historical characteristics of the previous tumor of the inguinal region, confirming the metastasis.
At present, the patient was referred to Mexico city for palliative radiotherapy sessions, pending to assess the response to it.
Discussion
Primitive neuroectodermal tumors grouped in the Ewing sarcoma family, are aggressive tumors, with poor prognosis that mainly affect pediatric age or young adult, rare in patients under 5 years or over 45 years of age, therefore, the presence of said tumor outside the ranges of age documented in the literature is rare1.
The main form of their presentation involves the bone system, and less common, the extraosseous presentation, among which are soft tissues, the inguinal region being a rare site for the location of these tumors2,3.
The presented case is of a 47-year-old female patient with histopathological diagnosis compatible with a primitive neuroectodermal tumor, with an unusual presentation in the left inguinal region, incidentally diagnosed after the gynecological examination due to an incomplete abortion because of an embryonic pregnancy3.
The diagnosis in the patient was made by the histopathological study compatible with the characteristic morphology of this family group of neoplasms, as well as an immunohistochemical panel compatible with this family of tumors, which presented positivity to CD99 and FLI-1, as literature mentions3,5.
The initial treatment was the surgical excision of the lesion in the inguinal area, followed by the radiotherapy sessions; the patient was not a candidate for adjuvant chemotherapy treatment due to deferral in the delivery of the results of the immunohistochemical techniques that were extended about 24 weeks after surgery. The treatment was adjusted to the patient conditions, receiving local treatment and with systemic treatment as a second instance, in case of recurrence.
The patient presents metastases at brain level in the right parietal area, a rare location, compared to lung, bone, and liver, among others that have a higher frequency5-7.
Conclusion
The neuroectodermal tumors continue to be an oncological problem due to their difficult therapeutic and their poor prognosis, however, it is a reality that each day, a little more is understood the physiopathology and oncogenesis of this family tumors, understanding more distinctly its genetic behavior and its clinical application. However, the etiology and biology of this type of tumors remain a field of study. As health personnel, it remains in us to favor the prognosis of this type of neoplasms with the timely detection and the correct multidisciplinary protocol.

