











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
The chordoma is a slow-growing1,2 rare malignant neoplasm and very destructive locally1-4. This tumor has received other names such as cordocarcinoma, chorioepithelioma, and notocordoma. It originates from remains of the notochord2 whose location can be found in the primitive deposit sites of the notochord as in along the axial skeleton, occipito-sphenoidal (clivus, 25%), vertebral (15%), and in the sacred region (60%). The age of incidence is very variable4,5. It can affect patients from infancy to the elderly. There is a critical point at 40 years2 as after this age, the 5-year survival forecast is notably lower. In general, it affects men more frequently4,5, especially those with an intracranial location5. Regarding their presentation in children, most are diagnosed around 10 years of age and chordomas are located intracranially5,6.
Symptoms depend on the location, affected intracavitary organs, and the sensory and motor condition1,7.
When the chordoma originates in the clivus, it can protrude toward the nasal cavity and present itself as a mass of central location4,5. They can have focal calcifications in up to 70% of cases, which can be detected with imaging studies8,5. When the chordoma involves the nasal passages, the differential diagnosis is very wide since there is great variety of tumors with different origins8. However, among the tumors that originate in the clivus are those formed by embryonic remnants.
Macroscopically, they are lobulated, shiny tumors with mucoid appearance2,4,5,7, which can occasionally present areas with hemorrhage, necrosis, and even cystic formations6. Histologically they are composed of two different cell populations. The so-called "boss cells" that are polygonal, elongated, eosinophilic, with a round to oval nucleus and epithelioid aspect that occasionally may exhibit certain pleomorphism. The other cell type is the so-called "physaliphorous cells"2,4-7,9 which are medium to large cells that contain cytoplasmic mucus Periodic Acid-Schiff (PAS) positive4. They have a vacuolated appearance, with small vesicles and some even have a "signet ring" appearance4. The nucleus is small and presents atypia that can vary from little apparent to very striking6,9. Atypical mitoses are rare10. All the cells are arranged in cords, groups, islands and even hepatoid cords suspended in a matrix of mucinous appearance, blue alcian, mucicarmine4, and PAS-positive, and occasionally lymphocyte inflammatory response is present. Three varieties of the chordoma are known: classic, chondroid, and dedifferentiated6,8,10,11. The chondroid variety, which is named after having an extracellular matrix of the hyaline cartilage type, occurs mostly in the cervical part and the clivus. The behavior of the three varieties is similar, although some authors mention that the chondroid variety is the least aggressive2. The latter occurs more frequently in patients younger than 40 years. It has also been mentioned that the dedifferentiated variety is the fastest growing and the most prone to metastasize8.
Characteristically, chordomas show hypodiploidy or pseudodiploid with structural rearrangements. The most frequent genetic anomalies are monosomy 1 and gain on chromosome 7. The locus 6q27, corresponding to the site of the growth factor Brachyura (growth factor T and specific marker of chordomas) is amplified in the cells of the chordoma. Receptors of growth factors such as Epithelial Growth Factor Receptor, α and β platelet-derived growth factor (growth factor derived from platelets), and c-KIT. Some tumor suppressor genes such as CDKN2A, CDKN2B, and FHIT6 have also been implicated in chordoma.
As already mentioned, the differential diagnosis of chordoma is very broad. Regarding the chondroid variety, chondrosarcoma should be ruled out. Immunohistochemical reactions are useful to determine the origin of the neoplasm. Among other types of tumors that can occur in the nasal cavities are carcinomas, both primary and metastatic, sarcomas, lymphomas, and melanomas, among others. When the location is in the sella turcica, radiologically, and clinically, it can be confused with pituitary adenoma1. It is worth mentioning that making these distinctions becomes even more difficult when the material of the biopsies is scarce10.
By immunohistochemistry, most chordomas are positive for cytokeratins AE1-AE3 and especially CK8, 18, and 196, epithelial membrane antigen (EMA)2,9,12, Brachyura10, PS100 as well as alpha1 anti-chymotrypsin. In some cases, they are positive for vimentin12, SOX-9, and lysozyme. Chordomas of chondroid type can be negative for cytokeratins and EMA2,12. The positivity for PS100 in this variant is > 80% compared to the other varieties2.
Chordoma treatment is surgical and involves the complete resection of the neoplasm. Frequently, it is complemented with radiotherapy or with a proton beam6. The complete resection of the neoplasm is difficult because the access to the point of origin is complicated3. The consequence of this is that there are recurrences of the tumor, and patients require surgery for the 2nd time. The incompletely resected chordomas can be associated with the development of sarcomas in both radiated and non-radiated patients7. The 5-year survival, in general, is 65%.
Presentation of the case
We present the case of a 48-year-old woman who began her condition 1 year earlier with bilateral nasal congestion with a predominance of the left side. Subsequently, he presented oppressive headache that was increasing from moderate to intense. A few days later, he presented paresis and paresthesias in the left side of the face. He went to the hospital center and on physical examination, paresis of the sixth cranial nerve was found with the consequent alteration in eye movements and hypermetry.
In the rest of the cranial nerve exploration, the following symptoms were found:
Decreased visual acuity
Anosmia
Anisocoria with left mydriasis
Paresis of the left side
Hypoesthesia of the left side
Paresis
Paresis
Without modifications
Without modifications
No alterations
Without modifications
Without modifications.
A few weeks later difficulty to swallowing, hypersalivation, and headache with increased intensity were noted. Imaging studies were carried out. Computed tomography showed an occupational lesion in the region of the clivus that extended to the nasal fossa (Fig. 1).
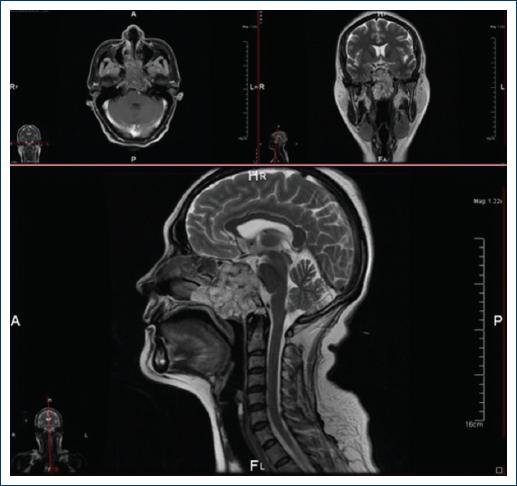
Figure 1 The images obtained by magnetic resonance are shown corresponding to axial, coronal and half skull cuts, weighted in T1 (top left) and T2 at the level of the Varolium and cerebellum bridge. A neoplastic lesion is identified that affects the spheno-occipital junction and extends to the ethmoid sinuses, is well delimited, homogeneous, and hypointense. At the top right at the level of the optic chiasm, the same neoplastic lesion that affects the floor of the skull is observed. This is an extra-axial lesion, heterogeneous, predominantly solid, hyperintense compared to brain tissue, has well-defined borders, compresses and displaces the right internal carotid artery. Below: half sagittal skull cut in which the aforementioned neoplastic lesion is observed that destroys the bone of the spheno-occipital junction dorsally compresses and displaces the Varolium bridge, and ventrally extends to the nasopharynx; it is a predominantly solid, homogeneous, nodular, hyperintense lesion with well-defined, pushing edges.
An incisional biopsy of the lesion was performed twice, but the material was insufficient for diagnosis. Both samples consisted of small fragments that together measured < 1 cm. A third biopsy was performed in which a neoplasm composed of two cell populations was identified. Some cells are found in distributed nests and cords. They are small, round, or oval with a moderate amount of cytoplasm and the nucleus is small. Other cells are large with vacuolated cytoplasm and show a vesiculated nucleus. Fibrosis bands and abundant mucoid material are identified among the neoplastic cells (Figs. 2-5). Finally, chordoma was diagnosed. Based on the diagnosis, radiation treatment was carried out. The treatment was concluded and the patient was operated 4 months later with the resection of most of the clivus tumor.

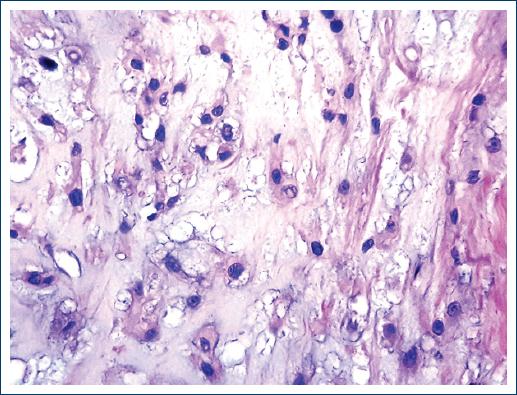
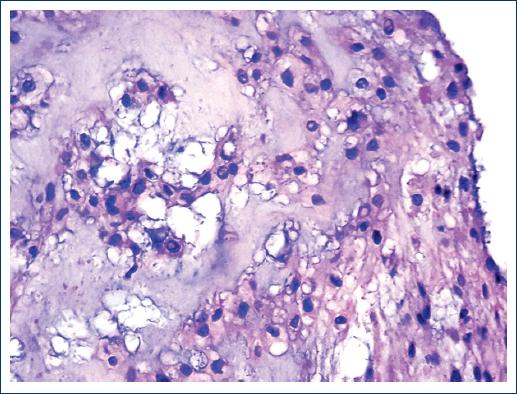

The piece consisted of several irregular fragments that together measured 4.5 × 3 × 1.5 cm. They were grayish white with a shiny surface, mucous appearance, and medium consistency. In some areas, the tissue had a cartilaginous appearance, and in others, there were pearly white papillary formations with a soft consistency.
Histologically, the neoplasm showed the same characteristics described in the previous biopsy.
The patient was discharged. Eight months later, she consulted the clinic for an intermittent discharge of clear fluid from both nostrils for more than 24 h. A diagnosis of the probable cerebrospinal fluid fistula was made, which was subsequently ruled out due to the disappearance of symptoms. At present, the patient is asymptomatic.
Discussion
Chordomas are neoplasms that are usually located along with the axial skeleton, predominantly in the region of the clivus and sacral region. The extension of the chordoma to the nasal cavity is not frequent that initially it manifests as a nasal tumor.
In a review, of 16 years (2000-2015) made in the archives of the pathology service of the General Hospital of Mexico decentralized agency (OD) and the UNAM School of Medicine, 28 cases of chordoma were found. Of these only five presented extension to the nasal cavity, including the present case.
Conclusions
Chordomas are rare neoplasms that are usually located at the end of the axial skeleton: clivus and sacral region. Its clinical manifestations are directly related to its location. The imaging studies combined with the clinical symptoms are definitive for diagnostic suspicion. The biopsy is essential to corroborate the diagnosis. The histological characteristics, although they are not pathognomonic, allow to discard the majority of the differential diagnoses.
The clinical diagnosis of a nasal tumor requires considering a large list of diagnostic possibilities. Taking into account the age of the patient and some specific data of the clinical manifestations, following diagnoses may be considered: lymphomas, carcinomas, melanomas, neuroblastomas, chondrosarcomas, and metastatic tumors are among the most frequent. The presentation of a chordoma in this location is exceptional; the reason why this case is published. Although the morphology of the neoplasm does not imply diagnostic difficulty, the location and its consequent manifestations are very rare.

