










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkVeterinaria México OA
On-line version ISSN 2448-6760
Veterinaria México OA vol.2 n.1 Ciudad de México Jan./Mar. 2015
Artículos científicos
Diversidad viral de comunidades de murciélagos en paisajes transformados de México
Oscar Rico Cháveza,b 0000-0002-9833-1101, Rafael Ojeda Floresa,b 0000-0002-7122-2968, Jesús Sotomayor Bonillaa,b 0000-0002-4756-8985, Carlos Zambrana-Torrelioc 0000-0002-5614-7496, Elizabeth Loza Rubiod 0000-0001-6812-9239, A. Alonso Aguirree 0000-0001-9507-857X, Gerardo Suzán*a,b 0000-0003-2508-6376
a Departamento de Etología, Fauna Silvestre y Animales de Laboratorio, Facultad de Medicina Veterinaria y Zootecnia, Universidad Nacional Autónoma de México, Avenida Universidad 3000, 04510, DF, México. *Autor para correspondencia: Gerardo Suzán. Tel: + 52 55-5622-5941
b Asociación Mexicana de Medicina de la Conservación, Kalaan-Kab A.C. Ciclistas 68-3, Col. Country Club 04220, DF, México.
c EcoHealth Alliance. 460 W 34th St., New York, NY 10001, U.S.A.
d Departamento de Biotecnología en Salud Animal. Centro Nacional de Investigación Disciplinaria en Microbiología Animal, Instituto Nacional de Investigaciones Forestales, Agrícolas y Pecuarias Km 15.5 Carretera Federal México-Toluca, Col. Palo Alto, 05110, DF, México.
e Department of Environmental Science and Policy, George Mason University, 4400 University Drive, Fairfax, Virginia 22030, U.S.A.
Recibido: 2014-11-13.
Aceptado: 2015-02-06.
Publicado: 2015-03-25.
Resumen
Mediante análisis epidemiológicos integrales, se estudiaron la diversidad beta (entre sitios) y la beta filogenética de sistemas multi-hospederos, así como los virus asociados con comunidades de murciélagos en paisajes fragmentados de Chiapas, Campeche y la zona metropolitana de la Ciudad de México. Se combinaron aplicaciones computacionales, técnicas de detección molecular y la secuenciación de nucleótidos de coronavirus, hantavirus, paramyxovirus y pegivirus con análisis ecológicos y filogenéticos.
Se descubrieron un total de 22 virus en 1 067 muestras pertenecientes a 42 especies de murciélagos, lo que representa una estimación de 78% de la riqueza viral total. Al aplicar el modelo estadístico Chao2 con datos de los 17 genotipos de virus encontrados con el mismo esfuerzo de muestreo, se calculó una riqueza viral de 23 genotipos. Usando un modelo con residuales, se categorizó a las especies hospederas y los tipos de hábitat que tienen mayor riesgo de asociarse con una mayor riqueza viral.
Se encontró una relación positiva entre la diversidad filogenética de los hospederos y la diversidad viral (r = 0.41, P < 0.05) y con la riqueza viral (r = 0.51, P < 0.05). La diversidad beta (tasa de recambio) de las comunidades de virus se explicó por la diversidad beta de los hospederos (r = 0.86, P < 0.05). Para entender los patrones de cambio en las comunidades virales y de hospederos, la diversidad beta se dividió en: un componente de anidamiento —pérdida de especies— y uno de recambio —disimilitud en la composición—.
En Chiapas, la diversidad beta de los hospederos se justificó por la anidación de especies, mientras que la diversidad beta filogenética se definió por el recambio de linaje de los hospederos. Campeche mostró altos valores de anidamiento filogenético y bajo recambio de hospederos. La diversidad beta y la diversidad beta filogenética indicaron que los patrones locales de la estructura de las comunidades de hospederos y las características abióticas regionales en paisajes dominados por actividades antropogénicas son un factor importante en la determinación de la composición de comunidades virales. Este estudio representa el primer esfuerzo en México para entender las relaciones hospedero-virus, a través del estudio de las relaciones entre la diversidad viral y las comunidades de murciélagos en paisajes transformados.
Palabras clave: Ecología de enfermedades; Riqueza viral; Diversidad alfa (diversidad de especies); Diversidad beta (diversidad entre sitios); Diversidad beta filogenética; Pérdida de hábitat; Chiroptera.
Introducción
Los cambios en el uso de suelo son el mecanismo principal que favorece la presencia de enfermedades zoonóticas (Patz et al., 2004). La expansión de la agricultura y la urbanización, de manera simultánea, han modificado la estructura y función de los ecosistemas, la estructura de las comunidades, los patrones de distribución de las especies y la biodiversidad (Christian et al., 2009; Gibbs et al., 2009). Estos sistemas modificados han creado ambientes idóneos para las interacciones multi-especie, en especial aquellos que involucran hospederos, vectores y/o patógenos (McMichael, 2004; Rivard et al., 2007; Jones et al., 2013; Rubio et al., 2014).
Se ha considerado a los murciélagos la principal fuente de agentes virales del tipo ácido ribonucleico (RNA) con alta patogenicidad como lyssavirus (Banyard et al., 2011), virus del Ébola (Leroy et al., 2005), virus de Marbug (Towner et al. 2009), virus Nipah (Epstein et al., 2006), virus Hendra (Smith et al., 2011) y coronavirus (CoV), por ejemplo: SARS, MERS (Hilgenfeld and Peiris, 2013).
Por otro lado, se reconoce a los murciélagos como un grupo clave para la conservación de los servicios ecosistémicos, pues sirven como polinizadores, dispersores de semillas y para el control de plagas en la agricultura (Medellín, 2009; Kunz et al. 2011). Además, debido a su manifiesta respuesta a la pérdida y fragmentación del hábitat, los murciélagos son excelentes bioindicadores de cambios en el ambiente (Medellín et al., 2000).
Para entender con propiedad las complejas interacciones en sistemas multi-hospederos, se realizaron varios análisis ecológicos y filogenéticos. En ecología de enfermedades, se han correlacionado los índices de diversidad, la riqueza (número de especies), la abundancia relativa de las especies en una comunidad (diversidad alfa) (Suzán et al., 2009) y los microbiomas (Anthony et al., 2013a; Olson et al., 2014), con la prevalencia de enfermedades. Los índices de diversidad también evalúan los cambios en la composición parásito–hospedero en comunidades de hospederos a escalas locales, regionales y biogeográficas —diversidad alfa, beta y gamma— (Svensson-Coelho y Ricklefs, 2011; Scordato y Kardish, 2014).
Desde la perspectiva evolutiva, se han estudiado las relaciones filogenéticas entre hospederos y patógenos, y se han incorporado índices de diversidad filogenética para medir cambios en las comunidades de hospederos a través de gradientes ambientales (Webb et al., 2002; Helmus et al., 2007). Dichos métodos filogenéticos ofrecen una dimensión adicional para explorar las interacciones entre hospederos y parásitos a través del tiempo, tales como especificidad de hospederos, co-evolución entre parásito y hospederos, eventos de cambio del hospedero y barreras filogenéticas en la transmisión de patógenos (Legendre et al., 2002; Streicker et al., 2010; Poulin et al., 2011).
El análisis ecológico y filogenético de las interacciones entre parásitos y hospederos puede integrar: la influencia del ambiente en la distribución de patógenos en escalas temporales y espaciales, así como diferentes niveles de organización biológica y taxonómica (Hawley y Altizer, 2011).
En esta investigación, se examinaron las relaciones entre la diversidad de hospederos y la de cuatro grupos virales asociados a murciélagos en paisajes dominados por actividades antropogénicas en México. Se pusieron a prueba dos hipótesis relacionadas con el efecto de la diversidad taxonómica y filogenética de hospederos sobre la diversidad viral e influencia del tipo de hábitat sobre la composición de comunidades de hospederos y de virus. En la primera se estableció que las comunidades de hospederos con alta diversidad taxonómica y filogenética soportarán una alta diversidad viral y en la segunda, los cambios en la composición de comunidades de virus y de hospederos en diferentes hábitats se reflejarán en los altos valores de diversidad beta y beta filogenética.
Material y métodos
Colecta de muestras
Los murciélagos se capturaron en tres sitios diferentes: Reserva de la biósfera Montes Azules (RBMA) en Chiapas, Reserva de la biósfera Calakmul (RBC) en Campeche, y la Ciudad de México y su zona metropolitana (GMC). Los primeros dos sitios están localizados en el sureste de México y representan regiones con una alta diversidad de especies, caracterizadas por grandes parches de vegetación primaria, mientras que GMC representa un sitio con una alta urbanización y algunos parches de vegetación. RBMA se caracteriza por bosque tropical perennifolio, mientras que en RBC domina una selva mediana subperennifolia; ambas regiones enfrentan una alta presión antropogénica.
Tanto en RBMA como en RBC los murciélagos se capturaron en tres hábitats: conservado (Fd), donde el impacto humano es casi imperceptible y persiste la vegetación primaria; fragmentado (F), caracterizado por áreas de vegetación primaria incrustadas en una matriz de agricultura y/o ganadería, y perturbado (D), zonas de transición entre áreas de vegetación secundaria y zonas agrícolas y/o ganaderas o urbanas. En GMC, los murciélagos se capturaron en el hábitat urbano (U) y fragmentado.
Se usaron cinco redes de niebla (9 m de ancho x 3 m de altura), por un periodo de 4 horas continuas desde el atardecer. Las especies se identificaron con una guía de campo (Medellín et al., 2008). La distancia mínima entre redes fue de 2 km en RBMA y de 10 km en RBC. Se llevó a cabo una "prueba de mantel" para asegurar la independencia por la distancia geográfica (RBMA, r = 0.55, P = 0.01; RBC, r = 0.57 P = 0.006). Se recolectaron muestras orales y rectales con hisopos, así como de sangre cuando fue posible, las cuales se transportaron en lysis buffer, preservadas a -80 ºC hasta el Center for Infection and Immunity de la Universidad de Columbia en Nueva York, EUA.
Detección viral
Se procesaron un total de 1 067 muestras de 608 individuos, pertenecientes a 42 especies de murciélagos para buscar cinco familias/géneros virales (cuadro S1). Se extrajo el ácido nucleico de todas las muestras con ayuda de la plataforma EasyMag® (bioMérieux, Inc Darham, NC, USA). Para la síntesis de cDNA se usó SuperScript® III first strand synthesis supermix (Invitrogen), según las instrucciones del fabricante. El reconocimiento viral se obtuvo mediante iniciadores consensos de PCR para el segmento L de hantavirus (HTV) (Klempa et al., 2006) y para el gen polimerasa (pol) para la detección de paramyxovirus (PMV) (Tong et al., 2008).
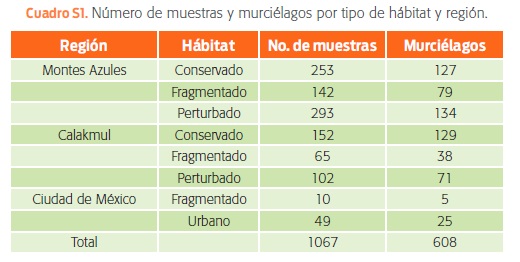
Los productos de PCR con el tamaño esperado se clonaron en StrataClone™ PCR cloning vector y se ordenaron de acuerdo con los iniciadores estándar M13R. Las detecciones de CoV, hepacivirus (HPV) y pegivirus (PGV) ya fueron previamente reportadas y sus secuencias virales pertenecen al mismo set de 1 067 muestras (Anthony et al., 2013b; Quan et al., 2013).
Estimación y cobertura de la riqueza viral
Se evaluó el esfuerzo de muestreo —número de muestras probadas para cada virus— con dos métodos: construyendo curvas de rarefacción y extrapolación, y mediante el cálculo de los valores residuales de un modelo de regresión lineal entre la riqueza viral dentro de un hospedero y el esfuerzo de muestreo por hospedero.
Las curvas de rarefacción y extrapolación son técnicas estadísticas para estimar el número de especies con base en un número de muestras (Magurran, 2004; Chao y Jost, 2012), lo que permite la evaluación del esfuerzo de muestreo y la determinación del número de muestras necesarias para obtener una riqueza viral con 95% de certeza (Chao et al., 2014).
Con la librería de R iNEXT (Hsieh et al., 2013), se evaluó la riqueza viral —definida como un único virus detectado en las 1,067 muestras— mediante la construcción de curvas de rarefacción y extrapolación basadas en un tamaño de muestra (3 201) tres veces mayor que el original (Chao et al., 2014).
Para identificar las especies hospederas asociadas con una mayor riqueza viral, se recurrió a la metodología propuesta por Herbreteau (2012). Se calcularon los valores residuales de una regresión lineal entre el logaritmo de la riqueza viral y el logaritmo del esfuerzo de muestreo por cada especie y por cada tipo de hábitat. Estos datos se transformaron en logaritmos para estabilizar la varianza. Se identificaron las especies y tipos de hábitat con mayor y menor riqueza con residuales positivos y negativos, según los esperados por este modelo de regresión.
Diversidad de hospederos y diversidad viral
Para conocer la diversidad de hospederos y de virus, se elaboraron matrices de abundancia, donde las filas representan los sitios y las columnas las especies o virus detectados. Con la librería R vegan (Oksanen et al., 2013), se calculó el índice de diversidad de Shannon-Wiener (Shannon, 1948) para cada matriz. Se reflejarán valores que variarán desde cero —cuando sólo existe una especie— hasta el logaritmo natural de la riqueza, cuando todas las especies se representan por igual (Magurran, 2004).
Diversidad filogenética y especificidad del hospedero
Para estimar la diversidad filogenética (PD) de las comunidades de hospederos se usó el súper árbol de mamíferos (Bininda-Emonds et al., 2007) con ayuda de la librería R Picante (Kembel et al., 2010). La PD se obtuvo al sumar el total del largo de las ramas del árbol filogenético de las especies muestreadas en cada hábitat (Faith, 1992). La relación entre la riqueza viral y la diversidad viral con la PD de los hospederos se exploró mediante un modelo lineal.
Debido a que los datos de la diversidad taxonómica no se distribuyeron de manera normal, sólo se analizaron con la información de PD. Para cuantificar las asociaciones hospedero–virus, se contrastó con un índice modificado de especificidad de hospedero, propuesto por Poulin y Mouillot, 2003, el cual mide la PD de las comunidades de hospederos asociadas a cada virus. Los virus con valores bajos de especificidad de hospedero muestran una alta plasticidad para infectar a un amplio rango de especies, mientras que los virus con valores altos están restringidos a especies con filogenias emparentadas (Poulin y Mouillot, 2003; Poulin et al., 2011).
Diversidad beta y diversidad beta filogenética
Para evaluar los cambios en la composición de comunidades virales y de hospederos dentro de cada región, se calcularon los componentes de BD: recambio espacial (ßSIM) y anidamiento (ßSNE) (Baselga, 2010). El recambio espacial mide la sustitución de las especies presentes por otras especies debido a factores ambientales o por aislamiento espacial, como la fragmentación del hábitat (Calderón-Patrón et al., 2012). El anidamiento determina si los sitios con un número de especies menor representan un subconjunto de los sitios con mayor riqueza de especies (Ulrich et al., 2009). Estos componentes se calcularon para poder realizar análisis de diversidad beta filogenética tanto a nivel taxonómico como filogenético.
La diversidad beta filogenética mide cómo las relaciones filogenéticas cambian en el tiempo y en el espacio, de la misma manera que la diversidad beta mide el cambio en la composición de especies a través del espacio (Graham y Fine, 2008). La PBD entre los niveles de perturbación se obtuvo con el inverso del índice de PhyloSor (Bryant et al., 2008). Este índice cuantifica las ramas compartidas de un árbol filogenético entre dos o más comunidades y adquiere valores de 0 —cuando no se comparten especies (ramas)— a 1, cuando en las comunidades están presentes las mismas especies.
Con la metodología propuesta por Leprieur (2012), se evaluaron los componentes de recambio filogenético (PßSIM) y anidación filogenética (PßSNE). Y de la biblioteca betapart implementada en R, se utilizaron las funciones beta.multi para estimar la BD y phylo.beta.multi para PBD. Así se conoció el peso de cada componente en el cambio de composición de las comunidades de virus y hospederos por tipo de hábitat en cada región estudiada (Baselga y Orme, 2013). Al final, mediante la correlación de Pearson, se exploró la relación entre la BD y la PBD de los hospederos con el cambio de composición de las comunidades virales por tipo de hábitat —obtenido mediante el índice de Sorensen.
Resultados
Comunidades de virus
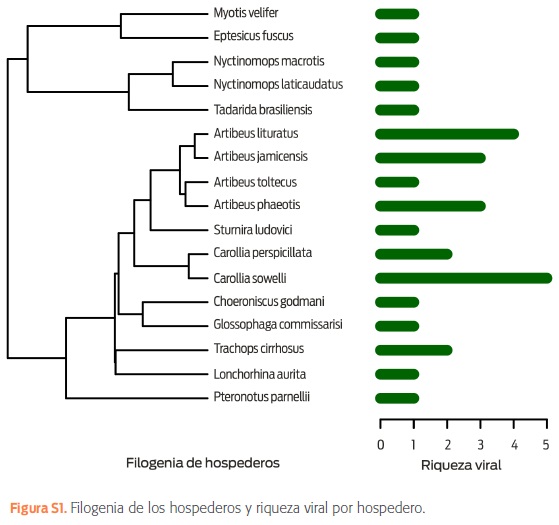
Se hicieron un total de 4,139 ensayos de PCR para la detección viral, incluyendo CoV (n = 1 067), PMV (n =1 067), HTV (n = 1 067), PGV (n = 469) y HPV (n = 469). La riqueza (S) fue de 22 genotipos virales en 46 muestras positivas de un total de 1 067 muestras: 13 para CoV, 2 para PMV, 2 para HTV y 5 para PGV (cuadro S2). No se detectaron HPV ni co-infecciones. Esta riqueza se asocia con 17 especies de murciélagos de 12 géneros y 4 familias (figura 1A). En hábitats conservados, se detectaron un total de 11 genotipos virales, seguidos de los fragmentados (10), perturbados (8) y urbanos (3) (figura 1B). Las especies con mayor riqueza viral fueron: Carollia sowelli (S = 5), Artibeus lituratus (S = 4), Artibeus jamaicensis (S = 3), Artibeus phaeotis (S = 3) y Trachops cirrhosus (S = 2), todas pertenecientes a la familia Phyllostomidae (figura S1).
Estimación y cobertura de la riqueza viral
Con base en los 17 genotipos virales detectados con el mismo esfuerzo de muestreo y el modelo estadístico Chao2 (Chao y Jost, 2012), se estimó una S de 23 genotipos. El esfuerzo de muestreo en las 1 067 muestras representa una cobertura de la riqueza estimada de 81%. La curva de rarefacción basada en un tamaño de muestra tres veces mayor (3 201 muestras) determinó una cobertura de 97% de la S (figura 2). El análisis por tipo de hábitat mostró una mayor cobertura, 53% en hábitats conservados, seguido de los sitios perturbados (43%) y los fragmentados (15%). La estimación de la riqueza viral por tipo de hábitat fue de 24 genotipos para sitios fragmentados, 19 para conservados y 10 para perturbados (cuadro 1).

Se observó una correlación positiva entre el esfuerzo de muestreo y la riqueza viral de los hospederos (R2 = 0.44, P < 0.01), y entre el esfuerzo de muestreo y la riqueza viral por tipo de hábitat (R2 = 0.37, P < 0.05). Sturnira lilium, Pteronotus parnelli y Artibeus jamaicensis se asociaron con una S mayor a la esperada por el modelo lineal entre el esfuerzo de muestreo y la S de virus en hospederos, mientras que Trachops cirrhosus, Lonchorhina aurita y Eptesicus fuscus se identificaron como especies con una S menor a la esperada (figura 3A). Comparado con lo supuesto en el modelo, el hábitat fragmentado de RBMA se asoció con una S mayor, mientras que el hábitat fragmentado de RBC se reconoció como el sitio con menor S (figura 3B).
Diversidad viral y de hospederos
En la región RBMA se detectó la mayor cantidad de virus (12), seguida de 11 en RBC y 3 en GMC (figura 4). Como se esperaba, RBMA fue la de mayor diversidad taxonómica y filogenética de hospederos, mientras el hábitat conservado fue el más diverso en ambas escalas (H = 2.79, PD = 484.8).
El hábitat perturbado mostró una mayor diversidad en ambas escalas, en comparación con el hábitat fragmentado (cuadro 2). A nivel regional, se observó una diferencia significativa entre la diversidad taxonómica (F = 13.63, gl = 2, P < 0.01) y la filogenética de hospederos (F = 16.71, gl = 2, P < 0.01), contrario a lo observado con la diversidad viral (F = 3.73, gl = 2, P > 0.05). Se detectó una correlación significativa entre la S y la diversidad viral con la diversidad filogenética de los hospederos. El 41% (P < 0.05) y el 51% (P < 0.05) de la varianza en la riqueza y en la diversidad viral, respectivamente, se explican por la diversidad filogenética de los hospederos. Este resultado sugiere que la composición de las comunidades de hospederos determina tanto la riqueza como la diversidad viral.

Especificidad filogenética del hospedero
De los 22 genotipos virales detectados, sólo seis se asociaron con más de dos especies de murciélagos (cuadro S3). El flavivirus PgV fue el virus con menor especificidad de hospedero (114.5), detectado en tres especies pertenecientes a tres subfamilias de flostómidos; Stenodermtinae y Phyllostominae, ambas de capturas en sitios conservados, además de Glossophaginae del hábitat perturbado. Se detectó el virus Hanta 2 (87.8) en dos especies de diferentes hábitats; Carollia sowelli (perturbado) y Trachops cirrhosus (conservado), mientras que el coronavirus MexCoV 5b (85.8) se encontró en tres especies, dos de las cuales son del mismo género: Artibeus jamaicensis, A. lituratus (conservado) y C. sowelli (fragmentado). Y se halló el MexCoV 1 (65.6) en dos especies del mismo género (figura 1A).
Diversidad beta
Se encontró que 86% (P < 0.05) de la BD de las comunidades virales se explicó por el recambio de hospederos entre hábitats. Se presentó la misma tendencia entre la PBD y la BD de las comunidades de virus, aunque el resultado no fue estadísticamente significativo (r = 0.79, P > 0.05). En el caso de RBMA, la disimilitud en la composición de las comunidades de hospederos a través de los hábitats fue relativamente elevada (ßSOR = 0.49), lo cual se explica por el componente de anidación (ßSNE = 0.57), es decir, las especies muestreadas en los hábitats menos ricos también están contenidas en el más rico (Fd = 26 especies).
La PBD mostró un comportamiento diferente, aunque el valor de disimilitud fue parecido (PßSOR = 0.42). El componente de recambio (PßSIM = 0.25) explicó parcialmente el cambio en la composición filogenética, debido a la baja relación filogenética en el subconjunto de especies que la anidación reemplazó. La diversidad beta total en RBC fue baja (ßSOR = 0.37), lo que sugiere que el tipo de hábitat tiene poca influencia en la estructura de las comunidades de especies.
El patrón de la diversidad beta en RBC fue diferente del de RBMA, si bien el componente de recambio (PßSIM = 0.22) influyó moderadamente en la disimilitud en la composición de las especies. El análisis de PBD mostró un alto reemplazamiento por especies en estrecha relación filogenética (PßSNE = 0.79). La interpretación de los patrones de BD en GMC es limitada, porque sólo se sometieron a muestreo dos comunidades, lo que hace imposible el cálculo del componente de anidación. Tanto en RBMA como en RBC se observaron altos valores de BD en las comunidades virales (cuadro S3), que se explicaron por el componente de recambio (ßSIM = 0.66). Esto sugiere que los cambios en la calidad del hábitat conducirán a un alto reemplazo de genotipos virales, independientemente de la composición de los hospederos.
Discusión
En este estudio se evaluó la relación entre la diversidad de murciélagos y la diversidad de cuatro virus de importancia médica dentro de un gradiente de tipo de hábitat en un paisaje dominado por actividades humanas en el sureste de México.
Gracias al uso de aplicaciones computacionales, técnicas moleculares para la identificación de virus y análisis ecológicos y filogenéticos, se cuantificó el recambio en la composición de comunidades virales asociadas a comunidades de murciélagos. Se encontró una fuerte relación entre la riqueza y la diversidad viral con la diversidad filogenética de los hospederos.
Especies generalistas se asociaron con una mayor cantidad de virus de lo esperado; además, se observó una relación positiva entre la diversidad beta tanto de comunidades virales como de hospederos con el tipo de hábitat. Como se esperaba, se manifestó una correlación entre la S y la diversidad de virus con la PD de los hospederos, lo cual sustenta dos hipótesis:
1. La hipótesis sobre el hábitat heterogéneo (Lawton, 1983), que propone una fuerte relación entre la diversidad ambiental –en este caso, la diversidad filogenética de los hospederos–, y la diversidad biológica (diversidad viral).
2. La hipótesis de estructuras clave (Tews, 2004), la cual se refiere a las especies hospederas que proveen de ciertos recursos a los virus como esas "estructuras clave" y que pueden estar vinculadas a diferentes especies virales.
Si se considera a los murciélagos hospederos como hábitats, las estructuras clave podrían categorizarse según las características relacionadas con la reproducción y abundancia (potencial, reproducción, longevidad, gremio trófico y masa corporal), el potencial de transmisión (distribución, tamaño de refugio, rango de dieta) y la filogenia (distinción evolutiva y distancia filogenética).
Estos hallazgos sugieren que factores como la fragmentación y la pérdida de hábitat podrían determinar la estructura de las comunidades de especies, cuyo resultado serían áreas con mayor riesgo de presentar brotes de enfermedades zoonóticas, como lo proponen Gay et al., (2014), Kamiya et al., (2014) y Rubio et al., (2014). Se requiere de futuros análisis para identificar qué rasgos de los hospederos pueden determinar las estructuras de las comunidades virales, aunque una mayor diversidad viral no implica un mayor riesgo de salud pública; de hecho, la correlación entre la diversidad viral y la de hospederos sugiere que sólo en condiciones donde se transforma el hábitat (fragmentos y sitos perturbados) habría mayor probabilidad de riesgo de salud.
La especificidad filogenética del hospedero obtenida en este trabajo no sólo refleja el número de especies de murciélagos infectadas con un virus en particular, sino que ayuda a explorar las relaciones filogenéticas entre los hospederos. Los resultados revelan que pocos de los virus detectados tienen una elevada plasticidad de hospedero, como PgV y HTV 2. La mayoría de los virus detectados muestran una alta especificidad filogenética.
Debido a la calidad de los datos, no se puede concluir que los virus encontrados en una sola especie de murciélago sean exclusivos de ésta; sin embargo, los datos de los coronavirus mostraron una alta especificidad de hospedero según el género (Anthony et al., 2013b).
Los cambios en la composición de las comunidades de virus a través del gradiente antropogénico evaluado manifestaron una fuerte dependencia del recambio filogenético de hospederos; sin embargo, esta relación no fue estadísticamente significativa cuando se consideró la diversidad taxonómica.
Se reportan diferentes patrones de BD entre regiones. Mientras que RBMA se caracterizó por un proceso de anidamiento a nivel taxonómico y un recambio filogenético, debido a una alta diversidad, la BD de RBC se explicó por un anidamiento filogenético debido a una diversidad homogénea.
Como se esperaba, se encontraron altos valores de diversidad beta en las comunidades de virus, lo que sostiene la hipótesis de perturbación, donde los cambios en el uso de suelo modifican las dinámicas de los parásitos en especies en sistemas multi-hospederos y favorecen la transmisión entre especies diferentes (Murray y Daszak, 2013). Se ha propuesto que las transformaciones del hábitat favorecerán la exposición a patógenos en nuevos hospederos, en especial, en zonas con una alta diversidad e influirán en las tasas de transmisión entre especies (Lloyd-Smith et al., 2009; Brearley et al., 2013; Murray y Daszak, 2013).
Los análisis de BD en ambas escalas, taxonómica y filogenética, han probado ser una herramienta útil para entender si son los hospederos o los filtros ambientales los que determinan la composición de parásitos (Svensson-Coelho y Ricklefs, 2011; Scordato y Kardish, 2014).
No fue posible demostrar que la filogenia de los hospederos determina la composición de las comunidades virales debido a las limitaciones espaciales de la investigación. Se requieren más estudios para probar la relación entre la estructura filogenética de los hospederos y la diversidad beta de las comunidades virales en escalas espaciales biogeográficas o en comunidades separadas por barreras geográficas (proceso de vicarianza).
Conclusiones
Este estudio demostró que la calidad y las características abióticas del hábitat determinan la diversidad viral en paisajes dominados por actividades humanas. La riqueza y diversidad viral se explican por la diversidad filogenética de los hospederos.
El componente de recambio del hospedero representa una dimensión inexplorada con un alto potencial para estimar la diversidad viral, en especial en paisajes fragmentados. En esta investigación se integraron análisis de diversidad viral y de hospederos dentro de un gradiente de tipo de hábitat, y representa el primer esfuerzo en México para cuantificar el recambio de comunidades virales asociadas a murciélagos y para monitorear la riqueza potencial de virus en fauna silvestre y su relación con la biodiversidad, tema necesario para entender cómo cambios en la función de los ecosistemas y actividades antropogénicas promueven la aparición de enfermedades.
Financiamiento
Este trabajo fue financiado por la Agencia de Desarrollo Internacional de los Estados Unidos (USAID) a través del programa Emerging Pandemic Threats PREDICT, NIH-AI57158 (NBC-Lipkin), NIH NIAID R01 A1079231 (non-biodefense EID), DTRA.
Agradecimientos
Agradecemos al Consejo Nacional de Ciencia y Tecnología (CONACyT) y al Programa de Posgrado en Ciencias de la Producción y de la Salud Animal de la Universidad Nacional Autónoma de México. Un estimado reconocimiento a la Comisión Nacional de Áreas Protegidas (CONANP) y a Rafael Lombera, quien apoyó la investigación de campo en la Reserva de la Biosfera Montes Azules, así como a Rafael Ávila, quien coordinó el muestreo en la Ciudad de México.
También le damos las gracias al Grupo de Ecología de Enfermedades del Departamento de Etología, Fauna Silvestre y Animales de Laboratorio de la Facultad de Medicina Veterinaria y Zootecnia, Universidad Nacional Autónoma de México, y a Rodrigo Medellín, quien revisó el manuscrito final e hizo comentarios valiosos.
Conflictos de interés
Los autores estipulan que no tienen conflictos de interés.
Contribución de los autores
Oscar Rico Chávez y Rafael Ojeda Flores: Contribuyeron de igual manera al trabajo y comparten la primera autoría.
Oscar Rico Chávez: Realizó el trabajo de campo y los análisis ecológicos y filogenéticos y escribió el manuscrito.
Rafael Ojeda Flores: Realizó el trabajo de laboratorio y escribió en manuscrito.
Jesús Sotomayor Bonilla: Realizó el trabajo de campo y revisó el manuscrito.
Carlos Zambrana Torrelio y Alonso Aguirre: Coordinaron el proyecto.
Elizabeth Loza Rubio: Revisó el manuscrito.
Gerardo Suzán: Dirigió el proyecto y escribió y revisó el manuscrito.
1) Anthony SJ, Epstein JH, Murray KA, Navarrete-Macias, I, Zambrana-Torrelio CM, Solovyov A, Ojeda-Flores R, Arrigo NC, Islam A, Ali Khan S, Hosseini P, Bogich TL, Olival KJ, Sanchez-Leon MD, Karesh WB, Goldstein T, Luby SP, Morse SS, Mazet JAK, Daszak P, Lipkin WI. 2013a. A Strategy to estimate unknown viral diversity in mammals. mBio 4(5):e00598-13. DOI:10.1128/mBio.00598-13. http://mbio.asm.org/content/4/5/e00598-13.full [ Links ]
2) Anthony SJ, Ojeda-Flores R, Rico-Chávez O, Navarrete-Macias I, Zambrana-Torrelio CM, Rostal MK, Epstein JH, Tipps T, Liang E, Sanchez-Leon M, Sotomayor-Bonilla J, Aguirre AA, Ávila-Flores R, Medellín RA, Goldstein T, Suzán G, Daszak P, Lipkin WI. 2013b. Coronaviruses in bats from Mexico. The Journal of General Virology 94: 1028–1038. DOI:10.1099/vir.0.049759-0. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3709589/ [ Links ]
3) Banyard AC, Hayman D, Johnson N, McElhinney L, Fooks AR. 2011. Bats and lyssaviruses. Advances in Virus Research 79, 239–289. DOI:10.1016/B978-0-12-387040-7.00012-3. http://www.sciencedirect.com/science/article/pii/B9780123870407000123 [ Links ]
4) Baselga A. 2010. Partitioning the turnover and nestedness components of beta diversity. Global Ecology and Biogeography 19(Issues 1):134–143. DOI: 10.1111/j.1466-8238.2009.00490.x. http://onlinelibrary.wiley.com/doi/10.1111/j.1466-8238.2009.00490.x/full [ Links ]
5) Baselga A, Orme C. 2013. Betapart: an R package for the study of beta diversity. Methods in Ecology and Evolution 3(Issue 5):808–812. DOI:0.1111/j.2041-210X.2012.00224.x. http://onlinelibrary.wiley.com/doi/10.1111/j.2041-210X.2012.00224.x/full [ Links ]
6) Bininda-Emonds ORP, Cardillo M, Jones KE, MacPhee RDE, Beck RMD, Grenyer R, Price SA, Vos RA, Gittleman JL, Purvis A. 2007. The delayed rise of present-day mammals. Nature 446:507–512. DOI:10.1038/nature07347. http://www.nature.com/nature/journal/v446/n7135/full/nature05634.html [ Links ]
7) Brearley G, Rhodes J, Bradley A, Baxter G, Seabrook L, Lunney D, Liu Y, McAlpine C. 2013. Wildlife disease prevalence in human-modified landscapes. Biological Reviews of the Cambridge Philosophical Society 88(Issue 2):427–442. DOI:10.1111/brv.12009. http://onlinelibrary.wiley.com/doi/10.1111/brv.12009/full [ Links ]
8) Bryant JA, Lamanna C, Morlon H, Kerkhoff AJ, Enquist BJ, Green JL. 2008. Microbes on mountainsides: contrasting elevational patterns of bacterial and plant diversity. Proceedings of the National Academy of Sciences 105(Suppl1):11505–11511. DOI:10.1073/pnas.0801920105. http://www.pnas.org/content/111/20/7166.full.pdf+html [ Links ]
9) Calderón-Patrón JM, Moreno CE, Zuria I. 2012. La diversidad beta: medio siglo de avances. Revista Mexicana de Biodiversidad 83:879–891. DOI:10.7550/rmb.25510. http://www.scielo.org.mx/scielo.php?script=sci_arttext&pid=S1870-34532012000300034 [ Links ]
10) Chao A, Gotelli NJ, Hsieh TC, Sander EL, Ma KH, Colwell RK, Ellison AM. 2014. Rarefaction and extrapolation with Hill numbers : a framework for sampling and estimation in species diversity studies. Ecological Monographs 84(Issue 1):45–67. DOI:10.1890/13-0133.1. http://www.esajournals.org/doi/abs/10.1890/13-0133.1 [ Links ]
11) Chao A, Jost L. 2012. Coverage-based rarefaction and extrapolation: standardizing samples by completeness rather than size. Ecology 93 (Issue 12):2533–2547. DOI:10.1890/11-1952.1. http://www.esajournals.org/doi/abs/10.1890/11-1952.1 [ Links ]
12) Christian K, Isabelle LV, Frédéric J, Vincent D. 2009. More species, fewer specialists: 100 years of changes in community composition in an island biogeographical study. Diversity and Distributions 15 (Issue 4):641–648. DOI:10.1111/j.1472-4642.2009.00569.x. http://onlinelibrary.wiley.com/doi/10.1111/j.1472-4642.2009.00569.x/full [ Links ]
13) Epstein JH, Field HE, Luby S, Pulliam JRC, Daszak P. 2006. Nipah virus: impact, origins, and causes of emergence. Current Infectious Disease Reports 8:59–65. DOI:10.1007/s11908-006-0036-2. http://link.springer.com/article/10.1007%2Fs11908-006-0036-2#page-1 [ Links ]
14) Faith DP. 1992. Conservation evaluation and phylogenetic diversity. Biological Conservation 61(1):1–10. DOI:10.1016/0006-3207(92)91201-3. http://www.planta.cn/forum/files_planta/1_119.pdf [ Links ]
15) Gay N, Olival KJ, Bumrungsri S, Siriaroonrat B, Bourgarel M, Morand S. 2014. Parasite and viral species richness of Southeast Asian bats: fragmentation of area distribution matters. International Journal for Parasitology. Parasites and Wildlife 3(2):161–170. DOI:10.1016/j.ijppaw.2014.06.003. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4142259/ [ Links ]
16) Gibbs KE, MacKey RL, Currie DJ. 2009. Human land use, agriculture, pesticides and losses of imperiled species. Diversity and Distributions 15 (Issue 2):242–253. DOI:10.1111/j.1472-4642.2008.00543.x. http://onlinelibrary.wiley.com/doi/10.1111/j.1472-4642.2008.00543.x/full [ Links ]
17) Graham CH, Fine PVA. 2008. Phylogenetic beta diversity: linking ecological and evolutionary processes across space in time. Ecology letters 11(12):1265–1277. DOI:10.1111/j.1461-0248.2008.01256.x. http://onlinelibrary.wiley.com/doi/10.1111/j.1461-0248.2008.01256.x/pdf [ Links ]
18) Hawley DM, Altizer SM. 2011. Disease ecology meets ecological immunology: understanding the links between organismal immunity and infection dynamics in natural populations. Functional Ecology 25(Issue 1):48–60. DOI:10.1111/j.1365-2435.2010.01753.x. http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2435.2010.01753.x/full [ Links ]
19) Helmus MR, Savage K, Diebel MW, Maxted JT, Ives AR. 2007. Separating the determinants of phylogenetic community structure. Ecology letters 10(Issue 10):917–925. DOI:10.1111/j.1461-0248.2007.01083.x. http://onlinelibrary.wiley.com/doi/10.1111/j.1461-0248.2007.01083.x/full [ Links ]
20) Herbreteau V, Bordes F, Jittapalapong S, Supputamongkol Y, Morand S. 2012. Rodent-borne diseases in Thailand: targeting rodent carriers and risky habitats. Infection Ecology and Epidemiology 2:18637. DOI:10.3402/iee.v2i0.18637. http://dx.doi.org/10.3402/iee.v2i0.18637 [ Links ]
21) Hilgenfeld R, Peiris M. 2013. From SARS to MERS: 10 years of research on highly pathogenic human coronaviruses. Antiviral Research 100(Issue 1):286–295. DOI:10.1016/j.antiviral.2013.08.015. http://www.sciencedirect.com/science/article/pii/S0166354213002234 [ Links ]
22) Hsieh TC, Ma KH, Chao A. 2013. iNEXT: An R package for interpolation and extrapolation in measuring species diversity [unpublished manuscript] [ Links ].
23) Jones BA, Grace D, Kock R, Alonso S, Rushton J, Said MY, McKeever D, Mutua F, Young J, McDermott J, Pfeiffer DU. 2013. Zoonosis emergence linked to agricultural intensification and environmental change. Proceedings of the National Academy of Sciences of the United States of America 110(21):8399–8404. http://www.pnas.org/content/110/21/8399.full [ Links ]
24) Kamiya T, O'Dwyer K, Nakagawa S, Poulin R. 2014. Host diversity drives parasite diversity: meta-analytical insights into patterns and causal mechanisms. Ecography 37(Issue 7):689–697. DOI:10.1111/j.1600-0587.2013.00571.x. http://onlinelibrary.wiley.com/doi/10.1111/j.1600-0587.2013.00571.x/full [ Links ]
25) Kembel SW, Cowan PD, Helmus MR, Cornwell WK, Morlon H, Ackerly DD, Blomberg SP, Webb CO. 2010. Picante: R tools for integrating phylogenies and ecology. Bioinfomatics 26(11):1463–1464. DOI:10.1093/bioinformatics/btq166. http://bioinformatics.oxfordjournals.org/content/26/11/1463.long [ Links ]
26) Klempa B, Fichet-Calvet E, Lecompte E, Auste B, Aniskin V, Meisel H, Denys C, Koivogui L, ter Meulen J, Krüger DH. 2006. Hantavirus in African wood mouse, Guinea. Emerging Infectious Diseases 12(5):838–840. DOI:10.3201/eid1205.051487. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3374458/ [ Links ]
27) Kunz TH, Braun de Torrez E, Bauer DM, Lobova TA, Fleming TH. 2011. Ecosystem services provided by bats. Annals of the New York Academy of Sciences, 1223:1-38. Issue: The Year in Ecology and Conservation. DOI: 10.1111/j.1749-6632.2011.06004.x. http://onlinelibrary.wiley.com/doi/10.1111/j.1749-6632.2011.06004.x/pdf [ Links ]
28) Lawton JH. 1983. Plant architecture and the diversity of phytophagous insects. Annual Review of Entomology 28:23–39. DOI:10.1146/annurev.en.28.010183.000323. [ Links ]
29) Legendre P, Desdevises Y, Bazin E. 2002. A statistical test for host-parasite coevolution. Systematic Biology 51(2):217–234. DOI:10.1080/10635150252899734. http://sysbio.oxfordjournals.org/content/51/2/217.full.pdf+html [ Links ]
30) Leprieur F, Albouy C, De Bortoli J, Cowman PF, Bellwood DR, Mouillot D. 2012. Quantifying phylogenetic beta diversity: distinguishing between "true" turnover of lineages and phylogenetic diversity gradients. Plos One 7(8):e42760. DOI: 10.1371/journal.pone.0042760. http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0042760 [ Links ]
31) Leroy EM, Kumulungui B, Pourrut X, Rouquet P, Hassanin A, Yaba P, Délicat A, Paweska JT, Gonzalez JP, Swanepoel R. 2005. Fruit bats as reservoirs of Ebola virus. Nature 438:575–576. DOI:10.1038/438575a. http://www.nature.com/nature/journal/v438/n7068/full/438575a.html [ Links ]
32) Lloyd-Smith JO, George D, Pepin KM, Pitzer VE, Pulliam JRC, Dobson AP, Hudson PJ, Grenfell BT. 2009. Epidemic dynamics at the human-animal interface. Science 326(5958):1362–1367. DOI:10.1126/science.1177345. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3891603/ [ Links ]
33) Magurran AE. 2004. Measuring Biological Diversity. Oxford, UK: Blackwell Science Ltd. [ Links ]
34) McMichael AJ. 2004. Environmental and social influences on emerging infectious diseases: past, present and future. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences 359:1049–1058. DOI:10.1098/rstb.2004.1480. http://rstb.royalsocietypublishing.org/content/royptb/359/1447/1049.full.pdf [ Links ]
35) Medellín, RA. 2009. Sustaining transboundary ecosystem services provided by bats. In: López-Hoffman L, McGovern E, Varady R, Flessa K. (eds.) Conservation of Shared Environments: Learning from the United States and Mexico. Tucson, USA: University of Arizona Press. pp. 170–187. [ Links ]
36) Medellín R, Arita HT, Sánchez HO. 2008. Identificación de los murciélagos de México. Clave de campo. DF, México: Instituto de Ecología, UNAM-CONABIO. [ Links ]
37) Medellín R, Equihua M, Amin M. 2000. Bat diversity and abundance as indicators of disturbance in neotropical rainforests. Conservation Biology 14(Issue 6):1666–1675. DOI:10.1046/j.1523-1739.2000.99068.x. http://onlinelibrary.wiley.com/doi/10.1111/j.1523-1739.2000.99068.x/full [ Links ]
38) Murray KA, Daszak P. 2013. Human ecology in pathogenic landscapes: two hypotheses on how land use change drives viral emergence. Current Opinion in Virology 3(1):79–83. DOI:10.1016/j.coviro.2013.01.006. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3713401/ [ Links ]
39) Oksanen J, Blanchet GF, Kindt R, Legendre P, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H. 2013. Vegan: Community Ecology Package. R package version 2.0-10 http://cran.r-project.org/web/packages/vegan/index.html [access: November 4th, 2014] [ Links ].
40) Olson SH, Parmley J, Soos C, Gilbert M, Latorre-Margalef N, Hall JS, Hansbro PM, Leighton F, Munster V, Joly D. 2014. Sampling strategies and biodiversity of influenza a subtypes in wild birds. PLoS ONE 9(3):e90826. DOI:10.1371/journal.pone.0090826. http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0090826 [ Links ]
41) Patz JA, Daszak P, Tabor GM, Aguirre AA, Pearl M, Epstein J, Wolfe ND, Kilpatrick AM, Foufopoulos J, Molyneux D, Bradley DJ, Amerasinghe FP, Ashford RW, Barthelemy D, Bos R, Bradley DJ, Buck A, Butler C, Chivian ES, Chua KB, Clark G, Colwell R, Confalonieri UE, Corvalan C, Cunningham AA, Dein J, Dobson AP, Else JG, Epstein J, Field H, Furu P, Gascon C, Graham D, Haines A, Hyatt AD, Jamaluddin A, Kleinau EF, Koontz F, Koren HS, LeBlancq S, Lele S, Lindsay S, Maynard N, McLean RG, McMichael T, Molyneux D, Morse SS, Norris DE, Ostfeld RS, Pearl MC, Pimentel D, Rakototiana L, Randriamanajara O, Riach J, Rosenthal JP, Salazar-Sanchez E, Silbergeld E, Thomson M, Vittor AY, Yameogo L, Zakarov V. 2004. Unhealthy landscapes: policy recommendations on land use change and infectious disease emergence. Environmental Health Perspectives 112(10):1092–1098. DOI:10.1289/ehp.6877. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1247383/pdf/ehp0112-001092.pdf [ Links ]
42) Poulin R, Krasnov BR, Mouillot D. 2011. Host specificity in phylogenetic and geographic space. Trends in Parasitology 27(Issue 8):355–361. DOI:10.1016/j.pt.2011.05.003. http://www.sciencedirect.com/science/article/pii/S147149221100095X [ Links ]
43) Poulin R, Mouillot D. 2003. Parasite specialization from a phylogenetic perspective: a new index of host specificity. Parasitology 126(Issue 5):473–480. DOI:10.1017/S0031182003002993. http://journals.cambridge.org/download.php?file=%2FPAR%2FPAR126_05%2FS0031182003002993a.pdf&code=919662fcaf57bed7954f7b01a59f9543 [ Links ]
44) Quan PL, Firth C, Conte JM, Williams SH, Zambrana-Torrelio CM, Anthony SJ, Ellison JA, Gilbert AT, Kuzmin IV, Niezgoda M, Osinubi MOV, Recuenco S, Markotter W, Breiman RF, Kalemba L, Malekani J, Lindblade KA, Rostal MK, Ojeda-Flores R, Suzan G, Davis LB, Blau DM, Ogunkoya AB, Alvarez Castillo DA, Moran D, Ngam S, Akaibe D, Agwanda B, Briese T, Epstein JH, Daszak P, Rupprecht CE, Holmes EC, Lipkin WI. 2013. Bats are a major natural reservoir for hepaciviruses and pegiviruses. Proceedings of the National Academy of Sciences of the United States of America 110:8194–8199. DOI:10.1098/rspb.2014.2124. http://dx.doi.org/10.1098/rspb.2014.2124 [ Links ]
45) Rivard DH, Poitevin J, Plasse D, Carleton M, Currie DJ, Ward DH, Curriet DJ. 2000. Changing species richness and composition in canadian national parks 14(issue 4):1099–1109. DOI:10.1046/j.1523-1739.2000.98247.x. http://onlinelibrary.wiley.com/doi/10.1046/j.1523-1739.2000.98247.x/pdf [ Links ]
46) Rubio AV, Avila-Flores R, Suzán G. 2014. Responses of small mammals to habitat fragmentation: epidemiological considerations for rodent-borne hantaviruses in the Americas. EcoHealth. DOI:10.1007/s10393-014-0944-9. http://dx.doi.org/10.1007/s10393-014-0944-9
47) Scordato ESC, Kardish MR. 2014. Prevalence and beta diversity in avian malaria communities: host species is a better predictor than geography. The Journal of Animal Ecology 83(issue 6):1387–1397. DOI: 10.1111/1365-2656.12246. http://onlinelibrary.wiley.com/doi/10.1111/1365-2656.12246/full [ Links ]
48) Shannon CE. 1948. A mathematical theory of communication. System Technical Journal, 379–423.
49) Smith I, Broos A, de Jong C, Zeddeman A, Smith C, Smith G, Moore F, Barr J, Crameri G, Marsh G, Tachedjian M, Yu M, Kung YH, Wang LF, Field H. 2011. Identifying hendra virus diversity in pteropid bats. PLoS ONE, 6(9):e25275. DOI:10.1371/journal.pone.0025275. http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0025275 [ Links ]
50) Streicker DG, Turmelle AS, Vonhof MJ, Kuzmin IV, McCracken GF, Rupprecht CE. 2010. Host phylogeny constrains cross-species emergence and establishment of rabies virus in bats. Science 329(5993):676–679. New York, NY, USA. DOI:10.1126/science.1188836. http://www.ncbi.nlm.nih.gov/pubmed/20689015 [ Links ]
51) Suzán G, Marcé E, Giermakowski JT, Mills JN, Ceballos G, Ostfeld RS, Armién B, Pascale JM, Yates TL. 2009. Experimental evidence for reduced rodent diversity causing increased hantavirus prevalence. PLoS ONE 4(5):e5461. DOI:10.1371/journal.pone.0005461. http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0005461 [ Links ]
52) Svensson-Coelho M, Ricklefs RE. 2011. Host phylogeography and beta diversity in avian haemosporidian (Plasmodiidae) assemblages of the Lesser Antilles. Journal of Animal Ecology 80(issue 5):938–946. DOI:10.1111/j.1365-2656.2011.01837.x. http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2656.2011.01837.x/full [ Links ]
53) Tews J, Brose U, Grimm V, Tielbörger K, Wichmann MC, Schwager M, Jeltsh F. 2004. Animal species diversity driven by habitat heterogeneity/diversity: the importance of keystone structures. Journal of Biogeography 31(1):79-92. DOI: 10.1046/j.0305-0270.2003.00994.X. http://onlinelibrary.wiley.com/doi/10.1046/j.0305-0270.2003.00994.x/full [ Links ]
54) Tong S, Chern SWW, Li Y, Pallansch MA, Anderson LJ. 2008. Sensitive and broadly reactive reverse transcription-PCR assays to detect novel paramyxoviruses. Journal of Clinical Microbiology 46(8):2652–2658. DOI:10.1128/JCM.00192-08. http://jcm.asm.org/content/46/8/2652.full [ Links ]
55) Towner JS, Amman BR, Sealy TK, Reeder Carroll SA, Comer JA, Kemp A, Swanepoel R, Paddock CD, Balinandi S, Khristova ML, Formenty PBH, Albarino CG, Miller DM, Reed ZD, Kayiwa JT, Mills JN, Cannon DL, Greer PW, Byaruhanga E, Farnon EC, Atimnedi P, Okware S, Katongole-Mbidde E, Downing R, Tappero JW, Zaki SR, Ksiazek TG, Nichol ST, Rollin PE (2009). Isolation of genetically diverse Marburg viruses from Egyptian fruit bats. PLoS Pathog 5(7):e1000536. DOI:10.1371/journal.ppat.1000536. http://journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1000536 [ Links ]
56) Ulrich DH, Almeida-Neto M, Gotelli NJ. 2009. A consumer's guide to nestedness analysis. Oikos 118(Issue 1):3–17. DOI:10.1111/j.1600-0706.2008.17053.x. http://onlinelibrary.wiley.com/doi/10.1111/j.1600-0706.2008.17053.x/full [ Links ]
57) Webb CO, Ackerly DD, McPeek MA, Donoghue MJ. 2002. Phylogenies and community ecology. Annual Review of Ecology and Systematics 33:475–505. DOI:10.1146/annurev.ecolsys.33.010802.150448. http://www.annualreviews.org/doi/full/10.1146/annurev.ecolsys.33.010802.150448 [ Links ]
 Dirección para correspondencia:
Dirección para correspondencia:
Gerardo Suzán
Correo electrónico: orichvet@gmail.com
Nota
Este artículo puede ser consultado en versión completa en http://www.revistas.unam.mx/index.php/Veterinaria-Mexico

