











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
El cáncer renal ocupa el duodécimo lugar en frecuencia en el mundo1, con un incremento anual de hasta el 2% en las últimas décadas2. Presenta una mayor incidencia en países desarrollados, como los Estados Unidos y los países de Europa, con predisposición por el sexo masculino (1.5:1)1,2 y entre los 60 y 70 años de edad. En el 80-90% de los casos corresponden a una histología de células claras, un 10-15% son papilares (el 70% de tipo 1) y solo un 4-5% son cromófobos. Dentro de la histología papilar existen dos tipos diferentes según las características histopatológicas descritas por Delahunt y Eble3. El carcinoma papilar de tipo 2 es poco frecuente y se considera que presenta una evolución con peor pronóstico, a diferencia del tipo 1, alcanzando una sobrevida a 5 años del 45%4.
Se conocen numerosos factores de riesgo implicados en el desarrollo del carcinoma renal, pero no se han determinado factores independientes para el subtipo histológico, siendo cada vez más relevantes los aspectos hereditarios y ambientales.
El síndrome asociado a leiomiomatosis hereditaria, así como al cáncer de células renales, es un trastorno genético muy raro, el cual condiciona y predispone a los pacientes a presentar múltiples leiomiomas cutáneos, carcinomas de células renales y, en mujeres, leiomiomas uterinos. Está causado por una mutación heterocigótica de la línea germinal del gen supresor de tumores fumarato hidratasa5,6.
En la histología papilar se conocen alteraciones genéticas específicas de cada subtipo; sin embargo, no está disponible en toda la población la realización de un mapeo genético que ayude a demostrar su etiología y la relación con síndromes hereditarios.
Caso clínico
Varón de 32 años, con obesidad de grado II, antecedente de piloleiomiomatosis y de hipertensión arterial sistémica, tabaquismo positivo. Presentó hematuria total no formadora de coágulos sin condicionar retención aguda de orina, con remisión espontánea. La tomografía computarizada contrastada reveló un tumor dependiente del riñón derecho, heterogéneo, lobulado, irregular, de 179 × 147 × 165 mm, con realce hasta 120 UH; glándula suprarrenal izquierda de 57 × 43 × 20 mm, con realce hasta 80 UH (Fig. 1); tórax sin lesiones. Se realizaron nefrectomía radical derecha y adrenalectomía izquierda, con reporte histopatológico en el producto de la nefrectomía de carcinoma renal papilar de tipo II (Fig. 1), y en el producto de la adrenalectomía de adenoma suprarrenal. La tomografía computarizada de control posquirúrgico a los 2 meses no mostró datos de actividad tumoral. Al sexto mes de seguimiento se realizó una tomografía por emisión de positrones (PET) (Fig. 2) que evidenció una lesión en el segmento hepático VI con SUV (standardized uptake value) máximo de 7.8, con dimensiones de 18 × 13 mm, adenomegalias retrocavas, paracavas e interaortocavas, lobuladas, con SUV máximo de 14, de hasta 9 mm. Testículos con lesiones con incremento de metabolismo, predominio izquierdo con lesión de 15 × 13 mm, con SUV máximo de 8.6. El ultrasonido testicular (Fig. 3) evidenció lesiones hipoecoicas bilaterales, la mayor en el lado izquierdo, con ligero aumento del flujo a la aplicación de Doppler. Los marcadores tumorales fueron: alfa-fetoproteína 7.84, gonadotropina coriónica humana fracción beta < 1.20, lactato deshidrogenasa 155. Se realizó orquiectomía radical izquierda con reporte histopatológico de tumor de células de Leydig (Fig. 4) de 2 cm, con inhibina (+), calretinina (+), vimentina (+) y sinaptofisina (−), sin invasión linfovascular, y biopsia de lesión en muslo izquierdo con reporte de leiomioma (Fig. 5). La tomografía computarizada de tórax mostró múltiples lesiones de 2-5 mm de diámetro con realce al medio de contraste. En el abdomen se observan dos imágenes sólidas que presentan realce anular y necrosis central, de 60 × 50 mm, en segmento VII hepático, y de 45 × 30 mm en el segmento VI. El lecho quirúrgico derecho muestra imágenes hipodensas, redondeadas, de aspecto nodular, de 15 mm. La biopsia de la lesión hepática reporta necrosis, y la biopsia retroperitoneal presencia de células neoplásicas, sin poder establecer el tipo histológico. Se realizó tumorectomía y metastasectomía hepática, con reporte histopatológico de lesión hepática y retroperitoneal con metástasis por carcinoma papilar de tipo 2. Inmunohistoquímica con CD 10 positivo y CK 7 negativo (Fig. 6). En el último estudio de seguimiento no hay datos de actividad tumoral y actualmente se encuentra en vigilancia oncológica y manejo con sunitinib.
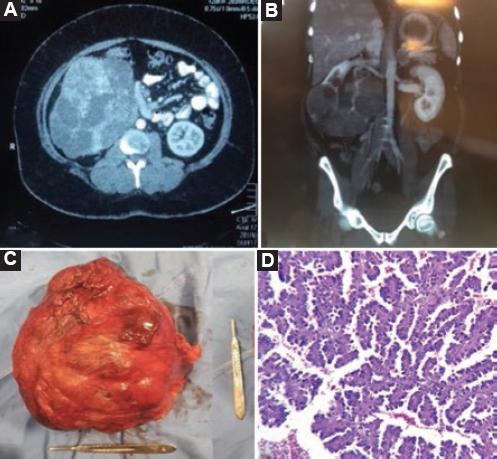
Figura 1 A: tomografía computarizada preoperatoria, corte axial. B: tomografía computarizada preoperatoria, corte coronal. C: pieza quirúrgica. D: tinción con hematoxilina-eosina; se observa una lesión con patrón de crecimiento papilar, con tallos fibrovasculares finos.
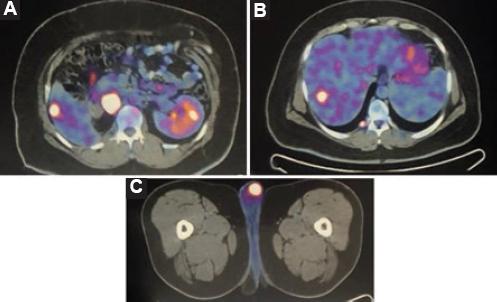
Figura 2 Tomografía por emisión de positrones con 2-fluoro-2-desoxi-D-glucosa. A: lesión hepática en el segmento VI, de 18 × 13 mm, con incremento del metabolismo, con SUV máximo de 7.8, lesión paracaval con SUV máximo de 14.0. B: lesion hepática. C: testículo izquierdo con lesión de 15 × 13 mm, con SUV máximo de 8.6.

Figura 3 Ultrasonografía testicular. A: testículo derecho con imágenes hipoecoicas con respecto al perénquima, la mayor de 0.94 × 0.80 × 0.79 cm, sin aumento importante del flujo en la aplicación del Doppler. Se observan otras lesiones pequeñas, de 0.53 cm aproximadamente. B: testículo izquierdo con ecos heterogéneos, lesión hipoecoica de 1.83 × 1.21 × 1.61 cm, aumento de la vascularidad al Doppler.

Figura 4 Producto de orquiectomía izquierda. A: corte con tinción de hematoxilina-eosina que muestra células poligonales de citoplasma amplio, eosinófilo, de límites imprecisos, con núcleo redondo central y nucleolo poco prominente. B: inmunohistoquímica, vimentina (+). C: inmunohistoquímica, inhibina (+).

Figura 5 Producto de biopsia de lesión en el muslo, corte con tinción de hematoxilina-eosina que muestra células fusiformes, alargadas, de músculo liso, profundas.
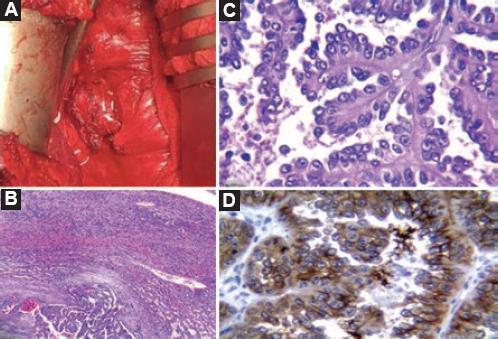
Figura 6 A: tumor paracaval de 7 × 5 × 3 cm, adherido firmemente a la cava en su porción medial. B: en la parte inferior se observa la lesión neoplásica que infiltra la pseudocápsula, y en la parte superior el parénquima hepático. C: papilas recubiertas por células neoplásicas grandes, con citoplasma eosinófilo, con núcleos hipercromáticos polarizados hacia el polo apical. D: inmunohistoquímica, CD10 (+).
Discusión
Existen muchos factores de riesgo asociados a los tumores renales, como causas genéticas, antecedentes familiares, tabaquismo, obesidad e hipertensión; actualmente se considera que los factores ambientales y de estilo de vida son tan importantes que podrían representar hasta el 20% de los casos de cáncer renal1. El cáncer renal es una enfermedad progresiva, de manera que hasta el 30% de los pacientes que al momento de diagnóstico están localizados presentarán metástasis a algún sitio en los primeros 5 años de seguimiento posquirúrgico4.
El carcinoma renal papilar corresponde al 10-15%2 de los pacientes con tumores renales, y de estos solo el 5% corresponden al tipo 2. La clasificación de Delahunt y Eble3 se implementó en 1997, describiendo características histológicas para cada tipo. Así, los de tipo 1 presentan papilas y estructuras tubulares, están cubiertos con pequeñas células que contienen citoplasma basófilo y a menudo tienen núcleos de bajo grado, mientras que los de tipo 2 son más heterogéneos, contienen papilas cubiertas por células grandes con citoplasma eosinofílico y muestran grandes núcleos de alto grado. Ambos tipos se han relacionado con alteraciones genéticas: en el tipo 1, con alteraciones en el oncogén MET (familiar), trisomías e incluso deleción Y, y en el tipo 2 con mutaciones del gen de la fumarato hidratasa (FH).
La leiomiomatosis hereditaria asociada a tumores renales es un síndrome resultante de la mutación del gen FH en el cromosoma 1q42.3-435,6. Se han identificado 11 mutaciones en el gen FH en el 76-93% de las familias con características clínicas compatibles con leiomiomatosis hereditaria y tumor renal6; es una enfermedad autosómica dominante. Los carcinomas de células renales asociados a leiomiomatosis familiar son agresivos, con el 70% de los pacientes muertos por enfermedad metastásica dentro de los 5 años posteriores al diagnóstico; además, se puede asociar al desarrollo de otros tumores poco comunes, los cuales están asociados con mutaciones en el gen FH, como son carcinomas de vejiga y próstata, cáncer de mama, tumores del estroma gastrointestinal, feocromocitomas, tumores de células de Leydig testiculares, adenomas suprarrenales y cistoadenomas ováricos7-9. La piloleiomiomatosis es un tumor benigno de músculo liso que se origina en los músculos erectores del folículo piloso, generalmente asintomático, que se asocia a dolor hasta en un 26% de los casos, y con afectación múltiple solo en el 4%10.
Ren, et al.4 demostraron que el tipo histológico y el estadio tumoral pueden considerarse factores de riesgo independientes para el pronóstico del carcinoma renal papilar, por lo que los pacientes con carcinoma papilar renal de tipo 2 que se encuentran en etapa pT3 o pT4 suelen tener una supervivencia general más corta en comparación con los que se encuentran en etapa pT1 o pT2.
En el seguimiento oncológico, el uso de la PET resulta limitado. Existen diversos marcadores que pueden utilizarse en el cáncer renal. Nakanishi, et al.11 realizaron un estudio en el que compararon el uso de PET con 2-fluoro-2-desoxi-D-glucosa (FDG) y 11C-colina para el seguimiento de pacientes con cáncer renal papilar, con una sensibilidad del 76% y del 88%, respectivamente, para establecer recurrencia, por lo que el empleo de la PET puede ser útil como procedimiento no invasivo para evaluar metástasis en todo el cuerpo y poder establecer la conducta terapéutica.
El tratamiento sistémico en los pacientes con enfermedad metastásica e histología de no células claras aún no está muy bien definido12. Se han evaluado diferentes fármacos angiogénicos, inhibidores de la tirosina cinasa y anticuerpos monoclonales, entre otros, que podrían mejorar la sobrevida global.
El estudio RAPTOR13 evaluó el everolimus como tratamiento de primera línea en pacientes con cáncer de células renales metastásico de tipo papilar y obtuvo una mediana de enfermedad libre de progresión de 7.9 meses para el tipo I y de 5.1 meses para el tipo 2, con una sobrevida global de hasta 17.8 mees para el tipo 1 y 20.5 meses para el tipo 2. Las medianas de tiempo libre de progresión y de sobrevida global fueron más altas en los pacientes con tipo 1 que en aquellos con tipo 2.
Un estudio de fase III se evaluó el temsirolimus frente al interferón y obtuvo un periodo libre de enfermedad de 4 vs. 1.7 meses, respectivamente, y una sobrevida global de 11.6 vs. 4.3 meses, respectivamente14.
En el metaanálisis de Fernández Pello, et al12; demostró una tendencia favoreciendo al sunitinib frente al everolimus, con una sobrevida libre de enfermedad promedio de 7.2 vs 4.9 meses y con una sobrevida global media de 31.6 meses con sunitinib y 10.5 meses con everolimus.
En un análisis multicéntrico realizado en Corea se evaluó bevacizumab + erlotinib en pacientes con leiomiomatosis hereditaria y carcinoma renal15. La mediana libre de progresión y la sobrevida global fueron de 13.3 y 14.1 meses, respectivamente, por lo que parece ser un tratamiento prometedor en pacientes con síndromes hereditarios.
Sin embargo, todos estos estudios no muestran diferencias significativas en la sobrevida libre de progresión entre los diferentes agentes empleados en tumores no de células claras. Por lo tanto, es importante continuar con el estudio de nuevos fármacos que mejoren el periodo libre de enfermedad y la sobrevida global.
Conclusiones
El carcinoma renal papilar de tipo 2 es poco frecuente. Su asociación con síndromes hereditarios con leiomiomatosis tiende a ser una enfermedad agresiva. Aún contamos con pocas herramientas terapéuticas que aporten buenos resultados; el sunitinib sigue siendo un fármaco que puede otorgar una respuesta favorable tanto en el carcinoma renal de células claras como en el de células no claras. Todavía falta mucho por avanzar en el tratamiento del cáncer renal, y es importante continuar con el estudio de nuevos fármacos que puedan mejorar la sobrevida de estos pacientes.

