











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
La ateroesclerosis se cataloga como una enfermedad vascular inflamatoria crónica, progresiva, con elevada participación inmunitaria1. Esta enfermedad se caracteriza por la acumulación de lípidos, especialmente la fracción conocida como lipoproteína de baja densidad (LDL), por debajo de la capa íntima de los vasos sanguíneos, convirtiéndose así en un potente estimulador de la respuesta inmunitaria en las células endoteliales vasculares2. Si bien el factor de riesgo más conocido y estudiado para el desarrollo de ateroesclerosis es la obesidad, este no es el único, ya que se ha descrito que la microbiota intestinal puede generar lípidos que desarrollen placas ateroescleróticas3. El papel que desempeñan las plaquetas en la ateroesclerosis, sobre todo en su desarrollo, aún no es comprendido del todo. La rotura de la placa y la subsecuente exposición de los componentes de la matriz extracelular permiten que las plaquetas se activen y desencadenen la formación del trombo, lo cual es frecuente en etapas avanzadas de esta patología; por lo tanto, las plaquetas no son simples mediadores de la hemostasia y la trombosis, sino que son un factor clave en el proceso inflamatorio característico de esta enfermedad1.
Lipoproteínas de baja densidad y su relación con la ateroesclerosis
Las LDL son consideradas como uno de los principales detonantes de la ateroesclerosis. Estos complejos macromoleculares formados por lípidos y proteínas participan en el metabolismo de los lípidos. Los estudios in vitro han mostrado que, independientemente de su concentración, las LDL en su forma nativa no se acumulan en los macrófagos; estas deben oxidarse primero para poder ser reconocidas por los receptores scavengers de dichos macrófagos para luego ser internalizadas por ellos. La oxidación de las LDL puede llevarse a cabo dentro o fuera de la capa íntima de los vasos sanguíneos4. En la circulación, las LDL pueden interacionar con radicales libres, lo cual conduce a su oxidación, misma que si se extiende por mucho tiempo da lugar a la formación de hidroperóxidos de ésteres de colesterilo, que se han propuesto como biomarcadores de estrés oxidativo. Las LDL, mediante transitosis, pueden pasar a la capa íntima del endotelio y oxidarse por diversos mecanismos:
- Oxidación mediada por células endoteliales, células del músculo liso o macrófagos, entre otras.
- Oxidación por peroxinitrito, el cual proviene del óxido nítrico (NO) que ha sido inactivado por el anión superóxido.
- Oxidación por proteoglicanos, que son abundantes en la matriz extracelular, sobre todo el sulfato de condroitina que puede unirse a la apolipoproteína B100 de las LDL.
- Oxidación por iones metálicos, como hierro o cobre, que reaccionan con enzimas como la mieloperoxidasa y la lipoxigenasa5.
-Oxidación por pH, el cual se ha descrito como ligeramente ácido en el centro de las lesiones ateroescleróticas, lo que favorece que las células endoteliales lleven a cabo la glucólisis anaerobia generando ácido láctico y provocando así la acidificación del espacio extracelular.
-Oxidación por glicación no enzimática de las LDL en la apolipoproteína B, en particular en el aminoácido de lisina.
Estos procesos favorecen la internalización de las LDL a los macrófagos y la polarización de estos hacia células espumosas, que son clave en el desarrollo de la ateroesclerosis4,6.
En la actualidad, las concentraciones de LDL (≥ 132 mg/dl) se utilizan como marcador de riesgo de padecer enfermedad cardiaca coronaria7. Los ácidos grasos procesados en el hígado (oxidados) son transportados por las lipoproteínas de muy baja densidad, también referidas como lipoproteínas ricas en triglicéridos, que a su vez intercambian estos triglicéridos con las lipoproteínas de alta densidad (HDL) y las LDL, lo que genera que ambas lipoproteínas se tornen ricas en triglicéridos para después perderlos en el hígado por la enzima lipoproteína lipasa hepática. Esto las convierte en LDL más pequeñas, más densas y más aterogénicas, mientras que las HDL, al también haberse tornado más pequeñas, pierden su capacidad protectora y además son catabolizadas más rápido en el riñón, lo que genera la baja de su concentración. Lo anterior conlleva que, aun cuando se mantengan las concentraciones adecuadas de LDL, haya un aumento del riesgo proaterogénico, causado por el tamaño y la pérdida de contenido de estas lipoproteínas, y por otra parte las concentraciones de triglicéridos aumentan en relación inversamente proporcional al diámetro de las partículas de LDL7-10.
Adhesión de las plaquetas al endotelio durante la ateroesclerosis
El tejido vascular es de suma importancia en el ser humano y es más que una capa de células que recubren los vasos; se considera un órgano metabólicamente activo que desempeña diversas funciones según el estímulo recibido11. Las plaquetas circulantes no solo participan en la rotura de la placa ateroesclerótica, sino que también potencian y modulan reacciones inflamatorias. De acuerdo con lo anterior, las plaquetas pueden adherirse a la pared de los vasos sanguíneos dañados o al endotelio activado o disfuncional, promoviendo el reclutamiento de células involucradas en la inmunidad innata y adaptativa1,12. El endotelio normalmente mantiene un estado antitrombótico por la liberación de diversas sustancias, como NO, prostaciclina y ciclooxigenasa 2 (COX-2); sin embargo, un endotelio activo cambia su estado a protrombótico aumentado la liberación de difosfato de adenosina (ADP) y de factor de Von Willebrand, e incrementando la expresión de factor tisular (TF), así como de diversas moléculas de adhesión, como las moléculas de adherencia intercelular (ICAM), las moléculas de adhesión a células vasculares (VCAM) y las proteínas quimiotácticas de monocitos 1 (MCP-1), entre otras12,13. La deposición de LDL nativa u oxidada en la capa íntima de las arterias promueve la activación del endotelio, lo que origina que las plaquetas circulantes se unan y activen14. Las plaquetas activadas independientemente del endotelio pueden unirse a este; por ejemplo, se sabe que las LDL oxidadas (LDLox) de la circulación se unen al receptor scavenger (CD36) de la plaqueta, el cual activa a NOX-2 (NADPH Oxidase Isoform 2) promoviendo la activación de estas y su consecuente unión al endotelio15.
La P-selectina es una molécula de adhesión indispensable para la unión de las plaquetas al endotelio a través del ligando de P-selectina (PSGL-1) o con el receptor del factor de Von Willebrand glicoproteína Ib (GPIb) presentes en el endotelio; a su vez, las plaquetas, mediante el mismo receptor GPIb, se unen a la P-selectina del endotelio, permitiendo la adhesión y el rodamiento de estas sobre la capa endotelial. Bajo altas fuerzas de estrés, las plaquetas activadas también pueden adherirse y rodar utilizando el mismo mecanismo13. Se ha observado que el receptor de colágeno GPVI, que se localiza en la superficie plaquetaria, se une al colágeno expuesto en la matriz subendotelial o al colágeno soluble, y este a su vez es reconocido por la integrina αvβ3 de la célula endotelial, lo que permite la adhesión y el rodamiento de las plaquetas sobre el endotelio13. Así mismo, el receptor GPIb que reconoce al factor de Von Willebrand se une a su receptor expresado en células endoteliales, lo cual también permite el rodamiento de las plaquetas sobre el endotelio (Fig. 1)13,16.
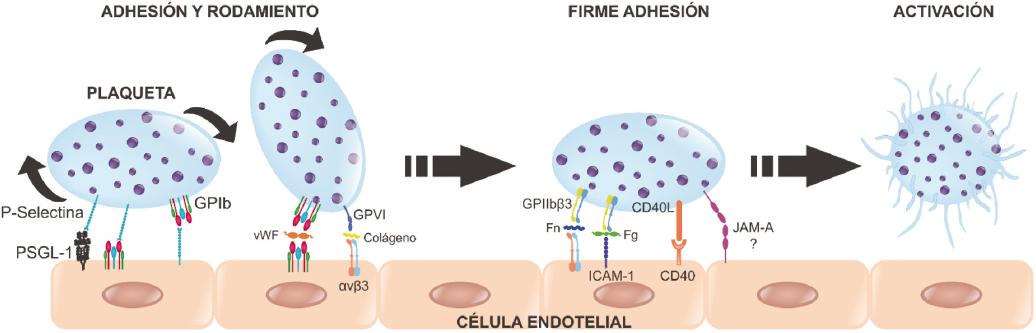
Figura 1 Activación plaquetaria. La activación de plaquetas es un proceso multietapa que comprende el acercamiento de estas al endotelio, la adhesión, el rodamiento y la firme adhesión. Durante dicho proceso, las plaquetas interactúan con las células endoteliales. La primera interacción es a través de la P-selectina en la plaqueta y la PSGL-1 en la célula endotelial, seguida de la interacción de la P-selectina de la célula endotelial y la GPIb de la plaqueta. El rodamiento requiere la unión de GPIb con GPIb en la plaqueta y la célula endotelial, mediada por el factor de Von Willebrand. La integrina GPVI de la plaqueta interactúa con la integrina αvβ3 de la célula endotelial, desencadenando a su vez en la plaqueta la activación de la integrina GPIIbβ3, que interacciona a través de la fibronectina con αvβ3 de la célula endotelial. También la ICAM-1 de la célula endotelial, a través del fibrinógeno, se une a la integrina GPIIbβ3 de la plaqueta. Finalmente, el CD40L de la plaqueta se une con el CD40 de la célula endotelial, promoviendo de esta forma la firme adhesión y la subsecuente activación plaquetaria. Fg: fibrinógeno; Fn: fibronectina; PSGL-1: ligando de P-selectina 1; vWF: factor de Von Willebrand.
Así mismo, los eventos antes descritos desencadenan la activación de las integrinas αIIbβ3 y αIIβ1, lo que genera agregación plaquetaria17. La integrina αIIbβ3 es capaz de unirse al fibrinógeno unido a ICAM-1 y a la fibronectina unida a αvβ3 en la célula endotelial, contribuyendo así a la adhesión estable18. La αIIβ1 puede unirse al colágeno que es reconocido por la integrina αvβ3 en la célula endotelial, lo que coopera a la adhesión estable, permitiendo que la plaquetas se activen (Fig. 1)13,19.
Otra proteína relacionada con la adhesión y la activación plaquetaria es el receptor 2 parecido a lectina tipo C, que activa vías de señalización que involucran a Src y PI3K, las cuales se ha observado que también se activan por los receptores antes mencionados (GPIb y GPVI)20.
Otra molécula que participa en la adhesión de las plaquetas al endotelio es el ligando de CD40 (CD40L o CD154), el cual, debido a la activación plaquetaria, forma complejos triméricos que se unen a su receptor CD40 de la célula endotelial, e incluso se ha descrito que pueden unirse a la integrina αIIbβ3 de otras plaquetas, lo que permite la formación de agregados. También se ha observado que el CD40L de las plaquetas se fragmenta y solubiliza formando CD40L soluble (sCD40L o sCD154), promoviendo la activación y la agregación de otras plaquetas y activando a las células endoteliales, que aumentan la expresión de moléculas de adhesión y la liberación de interleucinas y quimiocinas, generando también adhesión plaquetaria21 (Fig. 1).
La molécula de adhesión ocluyente A (JAM-A) de la plaqueta se creía que se unía a otra molécula JAM-A en la célula endotelial, y esto generaba activación plaquetaria, pero se ha observado que al unirse la JAM-A de la plaqueta a la JAM-A de otra plaqueta se genera inactivación de la integrina αIIbβ3, inhibiendo la agregación plaquetaria20.
Todas estas interacciones expuestas son solo algunas de las que pueden ocurrir durante la adhesión plaqueta-plaqueta y plaqueta-endotelio, cuya consecuencia es la activación plaquetaria con el fin de promover la formación del trombo o ejercer su función inmunitaria.
Mecanismos de activación plaquetaria
El factor de Von Willebrand se une a GPIbα, que forma parte del complejo receptor GPIb-IX-V de la plaqueta, induciendo que miembros de la familia de las cinasas Src fosforilen los motivos ITAM de GPIbα, lo que a su vez atrae a SYK (otro miembro de la familia Src), que fosforila a LAT22. La LAT fosforilada recluta a PI3K, que induce la activación de PLCγ2 y PKC, las cuales disparan la liberación de calcio del retículo endoplásmico23. El aumento del calcio induce la activación de Rap-1b, y la Rap-1b activada atrae a la talina, produciendo la activación de varias integrinas y en consecuencia la agregación plaquetaria24. La siguiente etapa de activación, que podemos llamarla de «amplificación», es dependiente de los agonistas derivados de los gránulos plaquetarios, tales como ADP, tromboxano A2 (TxA2), trombina, prostaciclina, serotonina o adrenalina, por mencionar algunos23. Dichos agonistas activan a los receptores acoplados a proteínas G, como P2Y1 y P2Y12 (receptores de ADP), PAR1 y PAR4 (receptores de trombina), TP (receptor de TxA2), 5-hidroxitriptamina (receptor de serotonina) y EP3 (receptor de prostaglandina E2)25. Estos receptores permiten la activación de PLCβ, que al igual que PLCβ2, tiene por objeto activar a PKC promoviendo el aumento intracelular de calcio y la activación de Rho, lo cual resulta en la activación de las plaquetas y la consecuente secreción de gránulos, aumentando la agregación de estas23,26.
El endotelio como órgano proateroesclerótico
El endotelio es considerado un órgano metabólicamente activo (autocrino y paracrino), que desempeña una función crítica en la regulación fisiológica del tono vascular, la adhesión, la migración celular y la resistencia a la trombosis, entre otras, todo ello en función del estímulo que recibe. Estas funciones las lleva a cabo mediante la liberación o la expresión de moléculas de adhesión, factores de crecimiento, compuestos antioxidantes y agentes vasodilatadores o vasoconstrictores. Para que se establezca la ateroesclerosis es necesario que el endotelio se active y se vuelva disfuncional. La activación se refiere a la capacidad de las células endoteliales para realizar nuevas funciones sin evidencia de lesión o disfunción celular. Así mismo, la disfunción endotelial surge al mantener las respuestas adaptativas de forma excesiva, sostenida y fuera de lugar, lo que altera la homeostasis vascular, predisponiendo así la pared vascular a la vasoconstricción, la adhesión leucocitaria, la activación plaquetaria, el estrés oxidativo, la trombosis, la coagulación o la inflamación, y favoreciendo el establecimiento de diversas patologías12,27,28.
En las células, en general, el NO es sintetizado a partir de L-arginina por un grupo de enzimas denominadas óxido nítrico sintetasas (NOS); en la célula endotelial, este proceso lo lleva acabo la óxido nítrico sintetasa endotelial (eNOS). En la ateroesclerosis, las concentraciones aumentadas y sostenidas de LDL en contacto con especies reactivas del oxígeno (ROS), sobre todo O2, permite su oxidación. La hiperlipidemia origina una regulación positiva de la NADPH oxidasa de la célula endotelial, lo que a su vez genera más ROS27. El aumento de este O2 no se puede inactivar por la superóxido dismutasa (SOD) y reacciona con el NO dando lugar a la formación de peroxinitrito, que a su vez es capaz de desestabilizar a la eNOS. Por otra parte, la dimetil arginina asimétrica (ADMA) compite con la L-arginina por el sitio de unión de eNOS, inhibiéndola29. También la LDLox aumenta la síntesis de caveolina-1, una proteína integral de membrana que igualmente inhibe a eNOS30. La inhibición de la eNOS trae como resultado un aumento de ROS (sobre todo O2) y una disminución del NO, lo que favorece la activación y la agregación plaquetaria (Fig. 2)30. Al generarse estrés oxidativo, este induce la desviación del estado redox normal hacia un ambiente oxidativo. El estado redox es el ambiente químico concerniente al número de equivalentes reductores disponibles, y uno de los principales es el sistema NADH/NAD+31-33.
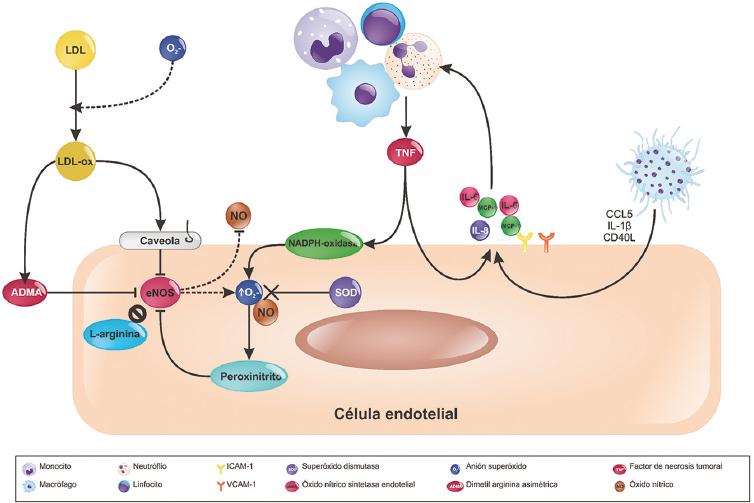
Figura 2 Disfunción endotelial en la ateroesclerosis. El aumento del anión superóxido (O2) interacciona con las LDL favoreciendo su oxidación. Las LDLox promueven un aumento de ADMA en sangre; este ADMA es un inhibidor competitivo de la eNOS, lo que conlleva la disminución en la producción de NO por la célula endotelial. Por otro lado, las LDLox favorecen la síntesis de caveolas en la célula endotelial, inhibiendo también a la eNOS. Aunado a esto, la estimulación positiva de la NADPH-oxidasa por el TNF secretado por células inflamatorias conduce a la sobreproducción del anión O2, lo que impide que la enzima SOD inactive a dicho O2 e interaccione con el NO dentro de la célula endotelial, dando lugar a la formación de peroxinitritos que inhiben a la eNOS. En conjunto con lo anterior, las plaquetas, mediante la secreción de CCL5, IL-1β y CD40L, pueden aumentar la expresión de moléculas de adhesión y la secreción de citocinas por parte de las células endoteliales, que actúan promoviendo el reclutamiento de células del sistema inflamatorio. ADMA: dimetil arginina asimétrica; eNOS: óxido nítrico sintetasa endotelial; LDL: lipoproteínas de baja densidad; NO: óxido nítrico; SOD: superóxido dismutasa; TNF: factor de necrosis tumoral.
La adhesión de las plaquetas al endotelio promueve que la plaqueta libere la quimiocina CCL5 (RANTES) y CD40L soluble, así como la expresión de CD40L, lo que conduce a la unión de las plaquetas a las células endoteliales, induciendo un aumento en la expresión de moléculas de adhesión y la liberación de citocinas y quimiocinas por la célula endotelial21. De igual forma, la liberación de interleucina (IL) 1β por las plaquetas induce que las células endoteliales liberen IL-6, IL-8, ICAM y MCP-1, entre otras, lo que también contribuye a su interacción con células inflamatorias (Fig. 2)34.
En etapas más avanzadas de la ateroesclerosis, el proceso inflamatorio en el que participan células de la inmunidad innata y adaptativa, como neutrófilos, macrófagos, células dendríticas y linfocitos, entre otros, genera un entorno rico en citocinas, que promueve la activación y la disfunción endotelial, y la unión y la activación de plaquetas al endotelio34. Así mismo, se ha observado que el estrés oxidativo amplifica la respuesta inflamatoria por parte de las células endoteliales que reclutan monocitos, linfocitos y neutrófilos, los cuales secretan citocinas como el factor de necrosis tumoral alfa (TNF-α), el cual, en la célula endotelial, puede regular positivamente a la NADPH oxidasa y negativamente a la eNOS, para de esta manera promover tanto la disfunción endotelial como el aumento de la expresión de citocinas, quimiocinas y moléculas de adhesión27.
Las lesiones ateroescleróticas avanzadas expresan un componente importante, denominado activador del plasminógeno tipo urocinasa (uPA), que es una proteasa en serina involucrada en la conversión del plasminógeno a plasmina y en el remodelado vascular35. Este uPA es expresado por las células endoteliales presentes en dichas lesiones, al igual que por macrófagos36. El uPA puede ser escindido en varios fragmentos biológicamente activos: fragmento amino terminal, dominio kringle y fragmento carboxi terminal, cada uno con propiedades únicas37. Por ejemplo, el fragmento amino terminal, al unirse al receptor del activador del plasminógeno tipo urocinasa (u-PARl), es capaz de inducir la migración de células del músculo liso vascular36, lo que desencadena la activación de diversas vías de señalización involucradas en la migración y la proliferación celulares, como MAPK y JAK-STAT38. Por otra parte, el uPA proveniente de células endoteliales de la zona de lesión y de células espumosas puede escindir, además del plasminógeno, otras proteínas de la matriz extracelular, degradar fibrina y activar a las metaloproteinasas (MMP), entre ellas la MMP-939. También el uPA promueve la formación de plasmina, la cual activa a la MMP-9 que degrada a la matriz extracelular ocasionando que la placa ateroesclerótica se torne vulnerable40-42. Por lo antes expuesto, se propone que el sistema uPA media la progresión de la ateroesclerosis43.
Interacción de plaquetas y leucocitos
Al igual que la adhesión plaquetaria al endotelio es mediada por la P-Selectina, la adhesión leucocitaria al endotelio y la formación de agregados plaqueta-leucocito se lleva a cabo por la interacción de la P-selectina con su ligando (PSGL-1)12. Sin embargo, se ha visto que los polimorfonucleares, en especial los neutrófilos, al secretar CCL5, y este al unirse a sus receptores CCR1 y CCR5, permiten que ingresen a la capa íntima de las arterias y liberen su contenido, como la azurocidina, que puede actuar sobre el endotelio promoviendo el aumento de expresión de moléculas de adhesión como ICAM-1, VCAM y E-selectina, catepsina G y CAMP (LL37), que promueven la atracción de monocitos hacia este44. Se ha observado que los neutrófilos pueden formar agregados plaqueta-neutrófilo, y estos responden a la LDLox aumentando su capacidad de transmigración hacia la capa íntima45.
El factor plaquetario 4, también llamado CXCL4, es una de las quimiocinas más abundantes presentes en los gránulos alfa de las plaquetas. Este CXCL4, junto con CCL5, se ha descrito que promueve la adhesión de monocitos al endotelio44. El CXCL4 promueve la polarización de macrófagos hacia un fenotipo denominado M4, que se caracteriza por tener baja capacidad fagocítica y alta expresión de transportadores de colesterol46.
En la formación de agregados monocito-plaqueta participan la PSGL-1/P-selectina y CD40/CD40L. Esta interacción provoca en el monocito la activación del factor nuclear kappa B (NF-κB), lo que a su vez induce un aumento de la expresión de proteínas inflamatorias, como MCP-1, TNF-α, IL-8 y Prostaglandina E2 (PGE2)47. La formación de agregados monocito-plaqueta se considera un marcador de activación plaquetaria con mayor valor predictivo incluso que la presencia de citocinas inflamatorias48.
Al unirse las plaquetas al endotelio por vía CD40L/CD40 se promueve el aumento de moléculas de adhesión, como ICAM-1 y VCAM-1, en la célula endotelial, así como la liberación de la quimiocina CCL2, lo que provoca el reclutamiento de linfocitos. La unión del linfocito a las plaquetas mediante PSGL1/P-selectina desencadena en el linfocito la activación de la integrina αL, provocando la adhesión de este a las células endoteliales mediante ICAM-149,50. Las plaquetas también promueven la adhesión de linfocitos T cooperadores a la matriz subendotelial dependiente de P-selectina y CD40L. También se ha documentado que, al unirse las plaquetas a los linfocitos B a través de CD40L/CD40, promueven en estos tanto su diferenciación como el recambio de clase de las inmunoglobulinas49.
Además, se ha descrito que la interacción de las células dendríticas con las plaquetas contribuye al desarrollo de la ateroesclerosis; por ejemplo, evidencias in vitro proponen que las plaquetas pueden reclutar células dendríticas mediante la interacción JAM-C/Mac-1 (antígeno-1 de macrófagos), promoviendo la fagocitosis de las plaquetas y la posterior muerte de las células dendríticas, induciendo de esta forma un ambiente inflamatorio51. Las plaquetas también pueden interactuar con células dendríticas por la unión CD40L/CD40, promoviendo la maduración y la diferenciación de las células dendríticas, además de estimular la liberación de citocinas y quimiocinas por parte de estas14,47.
Micropartículas derivadas de plaquetas
La mayoría de las células liberan vesículas extracelulares que derivan de su membrana; estas pueden contener diversas biomoléculas, como proteínas, lípidos, hidratos de carbono o ácidos nucleicos, entre otros, y tienen un impacto en células adyacentes o distantes. Las vesículas extracelulares fueron observadas originalmente por Chargaff y West, en 1946, como partículas procoagulantes derivadas de plaquetas; más adelante, en 1967, fueron referidas por Wolf como polvo plaquetario52. Estas vesículas contienen membrana proveniente de la célula de la cual se liberaron, y se clasifican de acuerdo con su función y origen celular (tolerosomas, prostatosomas, cardiosomas, vexosomas, etc.) o según su tamaño (exosomas de 40-120 nm; ectosomas, micropartículas o microvesículas de 100-1000 nm; cuerpos apoptóticos de 500-2000 nm). Las microvesículas o micropartículas derivadas de plaquetas son las más abundantes en la sangre humana y las más ampliamente estudiadas53. Estas micropartículas, en condiciones fisiológicas, tienen un papel en el mantenimiento de la homeostasis, pero bajo ciertos estímulos, como cáncer, enfermedades cardiovasculares, infección o inflamación, entre muchos otros, su número aumenta y se produce una desregulación de diversos sistemas, entre ellos el linfático y el de coagulación54. Estas micropartículas derivadas de plaquetas pueden contener caspasas (3 y 9), factores de transcripción (NF-κB, STAT, SOX), ligandos (CD40L, CXCL4), citocinas (IL-1β), quimiocinas (CCL5, CXCL7, CXCL4), factores de crecimiento Factor de crecimiento del endotelio vascular [VEGF], Factor de crecimiento derivado de plaquetas [PDGF], ácidos nucleicos (miRNAs, mRNAs), mediadores lipídicos (TxA2, ácido araquidónico), enzimas (COX-1, heparanasa) y restos de mitocondrias55. También estas micropartículas pueden ser procoagulantes debido a que su membrana es asimétrica en cuanto al contenido de fosfolípidos, como fosfatidilcolina y fosfatidilserina55. La fosfatidilserina expuesta en las micropartículas provee una superficie cargada negativamente, lo cual es requerido por los complejos protrombinasa y tenasa extrínsecos formados como parte del proceso de coagulación. Como se sabe, la coagulación inicia con la exposición del TF y la subsecuente unión de este al factor VII para formar el complejo tenasa extrínseco (TF-VIIa), el cual actúa sobre el factor X activándolo. Este factor Xa se une al factor Va en presencia de Ca+2 para formar el complejo protrombinasa (FVa-FXa). A su vez, el factor X también es activado por el complejo tenasa intrínseco (FVIIIa-FIXa) en presencia de Ca+2, y dicho factor Xa igualmente participa en la generación del complejo protrombinasa55,56. Estas mismas micropartículas derivadas de plaquetas también secretan inhibidor del activador del plasminógeno (PAI-1), lo que inhibe el sistema fibrinolítico; esto es importante sobre todo en etapas avanzadas de la ateroesclerosis, cuando la placa se vuelve susceptible a la rotura desencadenando el proceso de coagulación (Fig. 3)57.

Figura 3 Las micropartículas derivadas de plaquetas poseen fosfolípidos aniónicos que permiten el ensamblaje del complejo de iniciación de la coagulación (TF + FVIIa + Ca+2), además del complejo protrombinasa (FVa + FXa + Ca+2), lo cual conduce a un aumento de la trombosis. Las micropartículas también contienen integrinas, ligandos y moléculas de adhesión que permiten formar agregados con los leucocitos. Estos agregados de micropartículas y leucocitos interactúan con las células dendríticas y las células epiteliales. Por otra parte, estas micropartículas contienen citocinas y quimiocinas, otras moléculas derivadas de lípidos (TxA2 y 12-LO), enzimas y factores de crecimiento, entre otros, lo cual favorece el proceso inflamatorio. 12-LO: 12-lipoxigenasa; MP: micropartículas; PS: fosfatidilserina; TF: factor tisular; TxA2: tromboxano A2.
Debido a su contenido de caspasas, las micropartículas derivadas de plaquetas promueven la apoptosis de células endoteliales y leucocitos, y esto favorece que en las placas trombóticas haya un aumento de micropartículas tanto derivadas de plaquetas como de leucocitos y células endoteliales, mismas que se acumulan y potencian la formación del trombo y la ocurrencia de eventos inflamatorios58.
Las micropartículas derivadas de plaquetas pueden reclutar leucocitos, ya que poseen integrinas como αIIbβ3, GPIb, GPIX (CD42a) y GPIIIb (CD36), y moléculas de adhesión como P-selectina y PECAM-1, JAM-A, etc. Esto genera la adhesión de leucocitos a las micropartículas, formando agregados micropartícula-leucocito55. Estas micropartículas, al contener citocinas y quimiocinas, como IL-1β, RANTES y CXCL4, reclutan leucocitos hacia las células endoteliales, promoviendo la inflamación y la ateroesclerosis. Además, las micropartículas contienen factor activador de plaquetas, que permite la activación de neutrófilos y macrófagos (Fig. 3)55. Asi mismo, la LDL-ox puede interactuar con el receptor CD36 en la plaqueta, generando su activación y la subsecuente generación de micropartículas59.
Enfoques terapéuticos
Actualmente las estatinas son medicamentos de primera línea utilizados para el tratamiento de las concentraciones elevadas de lípidos plasmáticos, e incluso en la prevención primaria de enfermedades coronarias. Las estatinas actúan inhibiendo a la enzima 3-hidroxi 3 metilglutaril coenzima A (HMG-CoA) reductasa, la cual es la primera y principal enzima de la biosíntesis del colesterol; de esta manera se impide la generación de precursores de colesterol en el hígado y la reducción del colesterol LDL (considerado como uno de los principales factores de riesgo cardiovascular), ya que al reducir esta enzima se aumenta el número de receptores hepáticos para LDL (Fig. 4)60. Por otro lado, las estatinas tienen un efecto pleiotrópico muy amplio, pues pueden tener efectos antitrombóticos directos, como la simvastatina, que impide la interacción de la LDLox con monocitos y al mismo tiempo disminuye la expresión de TF inducido por LDLox sin causar un cambio en las concentraciones de colesterol61. A su vez, diversas estatinas se han implicado en disminuir la expresión del TF, incrementar la generación de trombomodulina e inhibir al factor V, entre otras acciones, lo que evita su activación y agregación. Además, se ha visto que la atorvastatina puede inhibir la activación plaquetaria directamente, mediante regulación negativa de la activación de la COX-162. Otras vías involucradas en inhibir la activación plaquetaria son la disminución del estrés oxidativo mediante la regulación negativa de NOX2 y de TxA2, y la liberación de NO; esto, por una parte, evita la activación y por ende la liberación de citocinas y quimiocinas, como CXCL4 y CXCL7, y por otro lado, la reducción del estrés oxidativo disminuye la capacidad de las LDL de ser oxidadas62,63. En conjunto con lo anterior, las estatinas pueden inhibir la fase de amplificación plaquetaria al inhibir la activación del receptor para trombina (PAR-4)64. Sumado a lo dicho, se ha demostrado que las estatinas (rosuvastatina) pueden disminuir el volumen plaquetario medio (VPM), el cual se considera como un posible marcador determinante de activación plaquetaria, debido a que las plaquetas circulantes en reposo tienen forma discoide, pero al activarse cambian de forma, ya que desarrollan filopodios y lamelipodios que generan un aumento de su superficie, mismo que se ve reflejado en un mayor volumen plaquetario medio65.

Figura 4 Tratamiento de la ateroesclerosis. Las estatinas inhiben la interacción de las LDLox con los macrófagos, lo cual impide la formación de células espumosas, y también bloquean la activación y la agregación plaquetaria. Los fármacos antiplaquetarios actúan inhibiendo la activación y la agregación plaquetaria mediante el bloqueo de diversos receptores a ADP, trombina y TxA2; además, algunos son capaces de inhibir la liberación de CD40L soluble y de IL-6, lo que potencia aún más su efecto antiplaquetario. Se han usado anticuerpos monoclonales como inhibidores de las integrinas αIIbβ3. LDLox: lipoproteínas de baja densidad oxidadas; TxA2: tromboxano A2.
Otros medicamentos que se usan ampliamente como tratamiento de las enfermedades coronarias son los antiplaquetarios, entre los que destacan tres: clopidogrel, prasugrel y ticagrelor. Los tres son inhibidores del receptor plaquetario de ADP (P2Y12), pero los dos últimos han demostrados ser más potentes y de actuación más rápida que el clopidogrel (Fig. 4)66. El clopidogrel se usa de manera estándar en combinación con aspirina como tratamiento antiagregante para reducir los eventos isquémicos en pacientes con enfermedad aterotrombótica; el clopidogrel, como ya se mencionó, es un inhibidor del receptor para ADP, mientras que la aspirina es un inhibidor irreversible de la COX-1, lo que resulta en una disminución de la producción de prostaglandinas y de TxA2, y con ello una reducción de la activación y la agregación plaquetaria67.
También se han desarrollado inhibidores del receptor para la trombina PAR-1, como vorapaxar y atopaxar; este último, además de inhibir dicho receptor en modelos múridos, ha demostrado disminuir la liberación de moléculas proinflamatorias, como CD40L soluble e IL-6, y la expresión de P-selectina. En humanos también demostró ser capaz de inhibir tanto la liberación de CD40L como el aumento de expresión de las moléculas de adhesión en la célula endotelial68.
El ticagrelor tiene un modo de acción único y diferente a los anteriores: no requiere activación metabólica para la actividad antiplaquetaria (a diferencia del clopidogrel y el prasugrel, que tienen un inicio de acción retardado al requerir activación metabólica) y se une de manera reversible a P2Y12, lo que permite la recuperación de la función plaquetaria más rápido (a diferencia del clopidogrel, que se une de manera irreversible a P2Y12)69.
De acuerdo con lo antes descrito, los efectos de los antiplaquetarios se basan en bloquear uno u otro receptor para los agonistas plaquetarios; sin embargo, como ya se ha comentado, las vías de señalización que cada receptor activa corriente abajo en algunos casos convergen entre ellas, mientras que en otros no, y esto es lo que lleva a utilizar terapias duales para tratar de abarcar la mayor cantidad de proteínas relacionadas con dichas vías y obtener el efecto esperado. A su vez, es necesario puntualizar que estos medicamentos tienen efectos permanentes, lo que podría generar algunos efectos adversos para ciertos pacientes. Por ejemplo, la terapia dual con aspirina y clopidogrel presenta la limitante de que el clopidogrel es un antiplaquetario irreversible, lo cual puede causar un riesgo significativo de hemorragia en pacientes que requieren cirugía y no han suspendido el tratamiento por 5-7 días. Otro efecto es la variabilidad de la inhibición plaquetaria por el clopidogrel, que explica por qué algunos pacientes, aun con dosis sostenidas de este fármaco, presentan reactividad plaquetaria elevada67. Debido a lo anterior han surgido nuevas versiones de estos bloqueadores de P2Y12 (receptor de ADP), como el cangrelor, que es un inhibidor directo y reversible de este receptor, y el elinogrel, que también actúa directamente sobre P2Y12 y es un inhibidor competitivo y reversible de este mismo receptor, y que al parecer tiene menos efectos secundarios que el clopidogrel70. Debido a lo complicado de la fisiología plaquetaria han surgido diversos intentos por mejorar la inhibición de las plaquetas y se ha intentado bloquear ciertas integrinas, como la αIIbβ3, con anticuerpos monoclonales. El abciximab es un anticuerpo quimérico ratón/humano que es capaz de inhibir la agregación plaquetaria al actuar directamente en estas integrinas71. La eptifibatida y el tirofibán también son antiagregantes al ser antagonistas de αIIbβ3. El primero es un heptapéptido cíclico de menos de 1 kDa, y un potente inhibidor de la unión del fibrinógeno a las plaquetas. El segundo es un péptido de bajo peso molecular y de corta acción, que en los humanos se une de manera reversible, y al parecer con baja afinidad, a αIIbβ372. Estos últimos medicamentos no se han estandarizado para uso humano, ya que no alcanzan a tener el efecto deseado en la mayoría de los pacientes y por ello se han relegado.
Conclusión
La ateroesclerosis es una enfermedad con características trombóticas e inflamatorias. El estudio de la ateroesclerosis en los humanos es limitado y generalmente se obtiene de disecciones de autopsias o de mediciones del aumento de alguna molécula en sangre como respuesta a un estímulo. Las plaquetas, que la mayoría de las veces solo se conocen por su participación en la trombosis, tienen una importante participación tanto en etapas tempranas de la ateroesclerosis como en etapas avanzadas, al haber rotura y una subsecuente exposición de matriz subendotelial. El papel de las plaquetas no se limita a la trombosis o la hemostasia, sino que tienen una participación activa en el proceso inflamatorio, ya que activan células como monocitos, neutrófilos, linfocitos y células dendríticas, entre otras, que favorecen un microambiente inflamatorio que ayuda al progreso de la ateroesclerosis. Las plaquetas tienen la capacidad de formar agregados y secretar citocinas y quimiocinas que actúan tanto sobre las células inflamatorias como el endotelio, promoviendo que este cambie a un estado proinflamatorio y proateroesclerótico. Estos procesos involucran diversas vías de señalización que desencadenan la activación de moléculas de adhesión, integrinas y receptores acoplados a proteínas G. Dichas vías han contribuido a desarrollar terapias farmacológicas, pero su complejidad ha impedido el desarrollo de fármacos más efectivos. El tratamiento antiinflamatorio para este padecimiento solo está representado parcialmente por las estatinas. Esto abre la posibilidad de tratar de inhibir o controlar el estado inflamatorio con nuevos esquemas de tratamiento, además del desarrollo de nuevos fármacos antiinflamatorios selectivos para la ateroesclerosis.

