










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista mexicana de ciencias pecuarias
On-line version ISSN 2448-6698Print version ISSN 2007-1124
Rev. mex. de cienc. pecuarias vol.10 n.3 Mérida Jul./Sep. 2019
https://doi.org/10.22319/rmcp.v10i3.4842
Articles
Variabilidad genética en una población de vacas Holstein utilizando marcadores SNP y su uso para monitorear estrategias de apareamiento
a Texas A&M University, College Station. Interdisciplinary Program in Genetics. Texas, USA.
b Texas A&M University. Department of Animal Science, Texas, USA.
cUniversità degli Studi di Milano. Department of Veterinary Medicine, Via Trentacoste 2, 20134 Milano, Italy.
dUniversità degli Studi di Milano. Azienda Agraria Didattico Sperimentale Angelo Menozzi, Landriano, Pavia, Italy.
e Universidad Autónoma de Querétaro. Facultad de Ciencias Naturales. Querétaro. México.
A medida que disminuyen los costos de la genotipificación, la demanda de ésta crece entre los ganaderos. En la mayoría de los hatos pecuarios es importante controlar el aumento del coeficiente de consanguinidad. Si bien esto es un motivo considerable para llevar a cabo la genotipificación, con frecuencia los productores desconocen los demás beneficios que puede traerles. El propósito de este estudio fue demostrar que es posible utilizar los chips SNP como una herramienta eficaz para el manejo de los hatos utilizando una población de ganado italiano Holstein-Friesian. Después de filtrar, se retuvo un total de 44 animales y 27,365 SNP para analizarlos. Los análisis de componentes principales (ACP) lograron identificar al progenitor y el efecto del origen del mismo en los análisis de componentes (ACP), y a la vez determinar que los sementales no influyen en los valores individuales del índice de selección genética. Los coeficientes de consanguinidad calculados a partir de los genotipos (F IS) permitieron tener un atisbo de la heterocigosidad del hato y determinar que la variabilidad genética se está manteniendo exitosamente. Por otra parte, se dedujeron los coeficientes de consanguinidad a nivel genético con base en los tramos de homocigosidad (F ROH) y se los comparó con los índices de consanguinidad basados en el pedigrí (F PED). Además, se identificaron 1,950 tramos de homocigosidad (ROH) con una longitud promedio de ROH de 4.66 Mb. Se caracterizaron los genes y QTL dentro de las regiones genéticas más comúnmente asociadas (primer 1 % y primer 5 % de los SNP). Estos resultados indican que la genotipificación de hatos pequeños, aunque a una densidad baja, facilita una mejor comprensión de la variabilidad genética dentro del hato y así, permiten a los productores gestionar su ganado desde una perspectiva genética.
Palabras clave Genotipos SNP; Holstein-Friesian; Consanguinidad; Tramos de homocigosidad; Manejo de rebaños
As genotyping costs continue to decrease, the demand for genotyping has increased among farmers. In most livestock herds, an important issue is controlling the increase in inbreeding coefficient. While this remains a large motive to genotype, producers are often unaware of the other benefits that genotyping could bring. The aim of this study was to demonstrate that SNP chips could be used as an effective herd management tool by utilizing a population of Italian Holstein-Friesian cattle. After filtering, the total number of animals and SNPs retained for analyses were 44 and 27,365, respectively. The principal component analyses (PCA) were able to identify a sire and origin-of-sire effect within the herd, while determining that sires do not influence individual genomic selection index values. The inbreeding coefficients calculated from genotypes (F IS) provided a glimpse into the herd’s heterozygosity and determined that the genetic variability is being well maintained. On the other hand, inbreeding coefficients on the genomic level were deduced from runs of homozygosity (F ROH) and were compared to the inbreeding coefficients based on pedigree (F PED). Furthermore, 1,950 runs of homozygosity (ROH) were identified with the average length of ROH being 4.66 Mb. Genes and QTL within the genomic regions most commonly associated (top 1% and top 5% of SNP) with ROH were characterized. These results indicate that genotyping small herds, albeit at low-density, provides insights to the genetic variability within the herd and thus allows producers the ability to manage their stock from a genetic standpoint.
Key words SNP genotypes; Holstein-Friesian; Inbreeding; Runs of homozygosity; Herd management
Introducción
Conforme disminuye el costo de la genotipificación, ésta tiene una demanda creciente entre los ganaderos. Los productores reconocen que es necesario conservar los recursos genéticos animales por su aportación al sustento humano, tanto en la actualidad como en el futuro1. El nivel de consanguinidad dentro del propio hato se ha convertido en una fuente de preocupación particular debido a los esfuerzos de selección, y por lo tanto ha sido un importante móvil para la genotipificación. El aumento de la consanguinidad tiene varios efectos negativos, como la reducción de los valores fenotípicos para obtener ciertos rasgos de la varianza genética, y una mayor frecuencia de genotipos homocigotos2.
Por ende, el deseo de gestionar este coeficiente ha crecido, sobre todo en los hatos locales pequeños o en el ganado lechero. No siempre se dispone de los pedigríes, o, si se tienen, estos no siempre bastan para calcular la consanguinidad dentro del propio hato; pero se puede determinar eficazmente el coeficiente de consanguinidad con base en los genotipos como un medio de lograr la heterocigosidad dentro de la población (F IS) o como una medida de consanguinidad genética total a partir de tramos de homocigosidad (ROH) (F ROH). De hecho, varios estudios en el ganado han demostrado eficazmente que los ROH son adecuados para estimar los coeficientes genéticos de consanguinidad en ausencia de un pedigrí3-5. En sí mismo el conocimiento de la homocigosidad proporciona nuevas posibilidades de administrar la consanguinidad en las especies de ganado y además puede ser utilizado para la asignación óptima de los recursos y el mantenimiento óptimo de la variación genética en razas bovinas intensamente seleccionadas6. No obstante, los ganaderos aún titubean en invertir en chips de polimorfismo de nucleótido simple (SNP) de alta densidad, por lo que recientemente se ha dado preferencia a los chips de baja densidad, puesto que son más rentables cuando se busca genotipificar a un gran número de animales. El diseño de estos chips ha mejorado mucho y ha dado lugar a mayores ganancias en términos de confiabilidad y de una mejor legibilidad entre los genotipos SNP7.
Otro importante motivo que los ganaderos tienen para genotipificar es que les permite estimar los valores genéticos de cría. De hecho, muchos criadores ya se basan en estos valores cuando adquieren semen7. Por ello, los ganaderos ya podían lograr tasas anuales de ganancia genética más altas mediante el uso de toros genéticamente probados; pero cada vez más se busca elevar el valor mediante la genotipificación de las hembras8. Se podría utilizar este valor adicional obtenido de la genotipificación del propio hato como una herramienta de gestión, puesto que los genotipos proporcionan mucha más información que sólo las variantes capturadas en el foco de la enfermedad que están presentes en el chip SNP. Este rango de información permite no sólo mejorar la confiabilidad de la selección genética (tanto de los toros como de las novillas) sino incluso identificar a las hembras selectas y a las mejores novillas para ser utilizadas en la reposición del ganado. Con ello los productores pueden obtener una mejor indicación del valor de un animal respecto al valor genético de cría esperado por sí solo y utilizarlo para evitar o gestionar la endogamia y, por supuesto, prevenir defectos genéticos evitando apareamientos que harían aflorar alelos perjudiciales8.
El objetivo de este estudio fue mostrar cómo se podrían utilizar los chips SNP como una herramienta eficaz de gestión de la variabilidad genética del hato. Se hace un énfasis particular en que el nivel efectivo de consanguinidad genética determinado con base en genotipos es comparable al que se obtiene mediante un pedigrí informativo, y que la genotipificación SNP permite revelar regiones genéticas bajo presión de selección en el hato identificado por genes con tramos de homocigosidad.
Material y métodos
Genotipificación
Los genotipos fueron proporcionados por la Asociación Nacional de Criadores de Frisona de Italia (ANAFI) para un total de 44 cabezas de ganado Holstein-Friesian de entre 12 y 15 meses de edad, provenientes todas de un solo hato, de la estación experimental de la Universidad. Todos los individuos tenían un índice de imputación de marcadores de más del 98 %. Los animales utilizados en este estudio fueron parte de una población experimental criada en la granja experimental de la Universidad de Milán, Azienda Agraria Didattico Sperimentale Angelo Menozzi. Los animales fueron genotipificados utilizando la versión 4 del arreglo GGP LD (GeneSeek®) para bovinos. Después de excluir los SNP que no estaban asignados a un cromosoma bovino BTA o que estaban asignados a BTA X o al ADN mitocondrial, quedaron 27,365 SNP.
Análisis de componentes principales (ACP)
La información adicional disponible para cada individuo incluía su pedigrí, su productividad genética, su funcionalidad, su índice de selección genética de tipo (GPFT), y su grupo de genotipificación. El ACP se llevó a cabo dentro de los SNP con el paquete informático SVS & Variation (Golden Helix®), versión 8.4 (SVS) (Golden Helix Inc., Bozeman, MT). El procedimiento utilizado en SVS tomó en cuenta 20 componentes principales, calculando para cada uno de ellos un valor de Eigen relativo: el primer componente principal (valor de Eigen = 1.188) fue graficado contra el segundo (valor de Eigen = 0.983).
Coeficientes de consanguinidad
Utilizando SVS se calculó el coeficiente de consanguinidad de F IS (o ƒ), definido como 1-(HETOBS/HETEXP). Este coeficiente es equivalente al índice de fijación dentro de las subpoblaciones de Wright con valores de entre -1 y +1. Todos los coeficientes de consanguinidad genética (F ROH) se calcularon según9:
Donde L ROH= es la longitud total de todos los ROH del genoma de un individuo, mientras que L AUTO se refiere a la longitud especificada del genoma autosómico cubierto por SNP (2,505,649,802 bp en el presente estudio).
Los coeficientes de consanguinidad basados en el pedigrí (F PED) se derivaron del programa Pedigree Viewer10. El pedigrí incluía registros de 2,760 individuos hasta 19 generaciones calculados por Pedigree Viewer, que ordena la información genealógica en las distintas generaciones. F PED y F ROH fueron comparados mediante la regresión lineal y el coeficiente de correlación de Pearson para probar su similitud y resaltar las diferencias entre los dos métodos de cálculo en un grupo de animales del mismo hato.
Tramos de homocigosidad
Se calculó el número de tramos de homocigosidad para cada individuo utilizando SVS, versión 8.4. Este programa no se basa en ventanas deslizables para identificar los ROH, sino que el algoritmo funciona de manera continua en todo un cromosoma, examinando todos los tramos posibles en busca de una correspondencia con los criterios especificados11. Se emplearon los siguientes criterios para definir ROH: 1) se permitía la ausencia de dos SNP; 2) se permitía un genotipo heterocigoto; 3) se estableció un número mínimo de 30 SNP como un tramo de homocigosidad entero, 4) una longitud mínima de 1 Mb y 5) un intervalo máximo de 1 Mb al siguiente SNP. Los tramos de homocigosidad se dividieron en cinco clases (<2, 2 a 4, 4 a 8, 8 a 16, y >16 Mb), utilizando la misma nomenclatura reportada por otros autores4,5. Se hizo un diagrama de la incidencia de tramos comunes por SNP empleando la herramienta Genome Browse de Golden Helix, y después se alinearon al ensamblaje bovino BTA U 5.0.1 para identificar los genes consistentes con el SNP en el primer 1% y el primer 5% de los tramos. La base de datos en línea STRING clasificó la red entre estos genes12, mientras se realizó la ontología de los genes (OG) mediante el sistema de clasificación Análisis de Proteínas mediante Relaciones Evolutivas (Protein Analysis Through Evolutionary Relationships, PANTHER) (Publicación 13.1), (http://www.pantherdb.org/pathway/).
Resultados
Análisis de componentes principales (ACP)
Los individuos se agruparon con base en su lote o grupo de genotipificación (No. 6), progenitor (n= 18), país de origen (n= 6) y caso de GPFT (n= 5). Los diversos lotes de genotipificación no se agruparon en absoluto, lo que indica que el tiempo de genotipificación no afectó a los genotipos individuales (no se muestran datos). Por el contrario, se ve una fuerte agrupación cuando se identifica a los individuos por su progenitor y por el país de origen de éste (Figura 1). Los progenitores 1 y 5 están particularmente distanciados de los demás, mientras que se tiende a agrupar a los originarios de Canadá, Italia y Estados Unidos, aunque por separado según su origen. Posteriormente se clasificó a los individuos por su valor de GPFT en incrementos de 500, y no se observó ninguna agrupación con base en esas categorías asignadas.

Figura 1 Diagrama de dispersión de los valores del análisis de componentes principales (ACP) basados en los genotipos SNP individuales. A) Valores del ACP agrupados por progenitor. B) Valores del ACP agrupados por origen del progenitor. C) Valores del ACP agrupados por Productividad, Funcionalidad, Clases de índice de selección genética de tipo (GPFT)
Coeficientes de consanguinidad
Se calculó la estimación desarrollada por Wright del coeficiente de consanguinidad F IS para cada individuo. La mayoría de los animales (n= 31) exhibieron un F IS negativo, mientras que la minoría (n= 13) tuvo coeficientes positivos (Figura 2). Los valores más altos y más bajos del F IS fueron 0.057 y -0.077, respectivamente. El F IS promedio se dio en -0.018, lo que provocó un aumento de la heterocigosidad debido a un esquema de exogamia utilizado por el ganadero. La mayoría de los individuos tuvieron coeficientes F PED de entre 0.050 y 0.070 (n= 31); los valores oscilaron entre 0.044 y 0.116. En su totalidad, la estimación del coeficiente de consanguinidad genética F ROH fue más alta que los coeficientes de consanguinidad calculados con base en el pedigrí. El F PED promedio fue de 0.064, mientras que el F ROH medio >1 Mb fue igual a 0.083.
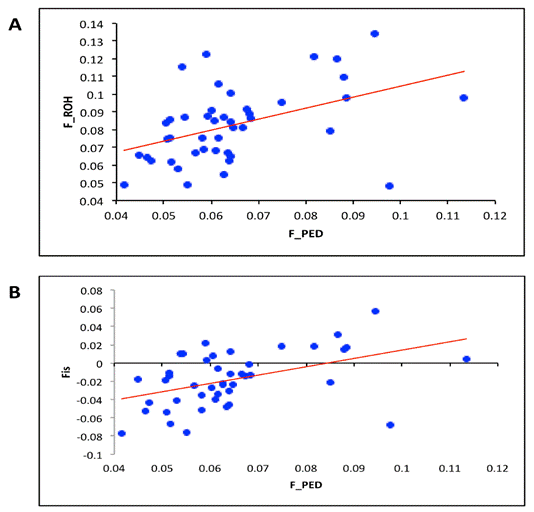
Figura 2 Regresión del coeficiente de consanguinidad individual. A) Regresión del coeficiente de consanguinidad calculado por pedigrí (F PED) sobre el coeficiente de consanguinidad calculado sobre el tramo de homocigosidad (F ROH). R2= 0.205; la línea roja indica la línea de regresión (F ROH = 0.042 + 0.621 * F PED ). B) Regresión del F PED sobre el F IS. R2= 0.214; la línea roja indica la línea de regresión. (F is= -0.077 + 0.913*F PED )
Tramos de homocigosidad
Habiéndose establecido el criterio de los ROH como de 1 Mb y un mínimo de 30 SNP, el software SVS identificó un total de 1,950 tramos dentro de la población, con una longitud de entre 1 y 22 Mb (Figura 3). Los 44 animales exhibieron ROH, con un número promedio de ROH por animal de 44. El número máximo de ROH para un individuo fue 64, mientras que el mínimo fue 25. La longitud promedio de los ROH fue 4.66 Mb, y la mayoría de los ROH se dieron en el rango de 4-8 Mb (Cuadro 1). BTA 10 tuvo el número máximo de ROH por cromosoma (122 tramos), y BTA 27, el mínimo (24 tramos). El ROH más largo se dio en BTA 10, con una longitud de 22 Mb, a diferencia de otros resultados que identificaron el ROH más largo en BTA 813-15. El número promedio de SNP que cayeron en una ROH fue consistente entre las categorías de longitud del ROH, pues osciló entre 42 SNP (<2 Mb) y 153 (>16 Mb). Al parecer, la longitud de los ROH fue relativamente proporcional al tamaño de los cromosomas, pues los tramos más largos se presentaron en los cromosomas más largos.
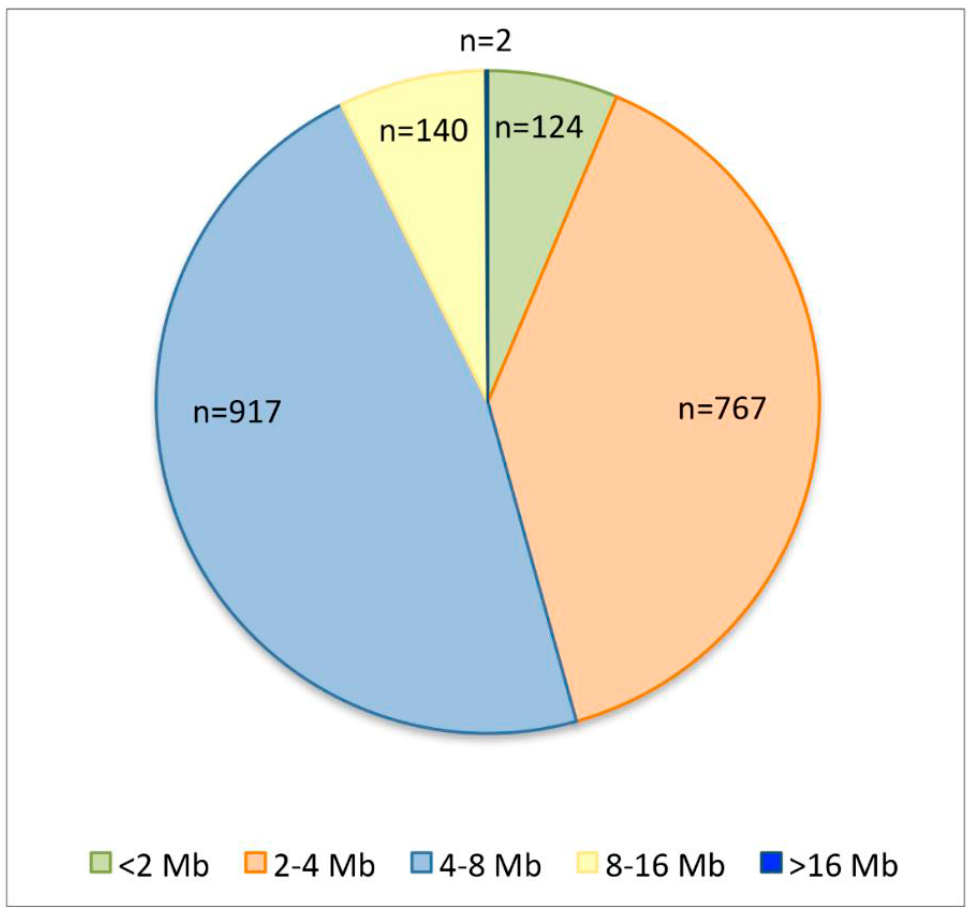
Cuadro 1 Números de ROH por cromosoma por clase de longitud de los ROH
| BTA | <2 Mb | 2-4 Mb | 4-8 Mb | 8-16 Mb | >16 Mb | Total |
|---|---|---|---|---|---|---|
| 1 | 1 | 20 | 71 | 4 | 0 | 96 |
| 2 | 1 | 31 | 75 | 10 | 0 | 117 |
| 3 | 5 | 22 | 27 | 6 | 1 | 61 |
| 4 | 1 | 37 | 47 | 7 | 0 | 92 |
| 5 | 7 | 36 | 40 | 15 | 0 | 98 |
| 6 | 11 | 48 | 45 | 2 | 0 | 106 |
| 7 | 3 | 45 | 51 | 7 | 0 | 106 |
| 8 | 5 | 32 | 56 | 10 | 0 | 103 |
| 9 | 10 | 23 | 29 | 0 | 0 | 62 |
| 10 | 2 | 43 | 64 | 12 | 1 | 122 |
| 11 | 2 | 36 | 41 | 6 | 0 | 85 |
| 12 | 5 | 14 | 16 | 4 | 0 | 39 |
| 13 | 8 | 32 | 39 | 6 | 0 | 85 |
| 14 | 8 | 50 | 21 | 0 | 0 | 79 |
| 15 | 3 | 20 | 22 | 4 | 0 | 49 |
| 16 | 12 | 25 | 34 | 7 | 0 | 78 |
| 17 | 2 | 17 | 21 | 3 | 0 | 43 |
| 18 | 5 | 21 | 14 | 2 | 0 | 42 |
| 19 | 7 | 30 | 16 | 1 | 0 | 54 |
| 20 | 2 | 24 | 43 | 7 | 0 | 76 |
| 21 | 1 | 24 | 26 | 6 | 0 | 57 |
| 22 | 2 | 20 | 20 | 5 | 0 | 47 |
| 23 | 4 | 16 | 12 | 2 | 0 | 34 |
| 24 | 4 | 16 | 21 | 3 | 0 | 44 |
| 25 | 1 | 22 | 9 | 0 | 0 | 32 |
| 26 | 1 | 21 | 11 | 2 | 0 | 35 |
| 27 | 3 | 12 | 7 | 2 | 0 | 24 |
| 28 | 1 | 13 | 18 | 4 | 0 | 36 |
| 29 | 7 | 18 | 20 | 3 | 0 | 48 |
| Total | 124 | 768 | 916 | 140 | 2 | 1950 |
Se identificaron las regiones genéticas más comúnmente asociadas a los ROH seleccionando los primeros 1 y 5 % de los SNP más frecuentemente observados (Figura 4, Cuadro 2). La incidencia de los tramos comunes por SNP indicó que la distribución genética de los ROH no fue uniforme en todos los cromosomas. Se identificaron los loci de caracteres cuantitativos (QTL) correspondientes con las regiones de homocigosidad que alberga el primer 5% de los SNP utilizando la Base de datos de loci de caracteres cuantitativos (QTL) de animales (AnimalQTLdb) (Cuadro 2)16.

Figura 4 Incidencia de SNP en ROH identificados con SVS. las líneas rojas y azules indican el límite adoptado para el 1% y 5% máximos, respectivamente
Cuadro 2 Regiones genéticas de la homocigosidad extendida en correspondencia con el primer 1 y 5% de los SNP
| BTA | Número de SNP |
Inicio (bp) | Final (bp) | Genes, | QTL (https://www.animalgenome.org/cgi-bin/QTLdb/BT/index) |
|---|---|---|---|---|---|
| 1 | 11 | 49891423 | 51147114 | HSF2BP, ALCAM, CBLB | Porcentaje de beta-lactoglobulina en la leche QTL (108870); Rendimiento de carne magra QTL (37225) |
| 1 | 18 | 145059132 | 146790949 | PDE9A, PKNOX1, ITGB2, ADARB1, POFUT2, ICOSLG, TRPM2, SIK1 | Porcentaje de proteína en la leche QTL (105990); Perfil de ácidos grasos en la leche C18 QTL (32646) |
| 2 | 11 | 15361703 | 17142680 | -- | Peso corporal (añal) QTL (66889); Puntuación de la ubre, QTL de Turgencia de la ubre (106708); Intervalo al primer estro después del parto QTL (30300) |
| 2 | 25 | 86178360 | 88823250 | PGAP1, ANKRD44, RFTN2, BOLL, PLCL1, SATB2, FBX036 | Rendimiento de grasa en la leche QTL (122473) |
| 2 | 28 | 119194847 | 132528595 | ECE1, ALPL, SLC16A14, SP140, SP110, SP140L, CAB39, PSMD1, SPOCD1, FABP3, SNRNP40, LAPTM5, HSPG2, USP48, RAP1GAP | Grosor de la grasa en la 12a costilla QTL (126458); Resistencia a las garrapatas QTL (135875); Rendimiento de grasa en la leche (desviación de las hijas) QTL (25782); Contenido de ácido lignocérico QTL (19771, 19709) |
| 3 | 36 | 101384522 | 105043063 | GPBP1L1, PRDX1, TESK2, ZSWIM5, PTCH2, KIF2C, C3H1orf228, RNF220, ERI3, SLC6A9, ST3GAL3, SLC2A1, PPCS, FOXJ3 | Peso corporal (al destete) QTL (24748); Índice de nacimientos QTL (15168); Tamaño del ternero QTL (15167, 15169); Facilidad para parir QTL (15170) |
| 5 | 25 | 55540165 | 56247946 | ANKS1B, AVIL, TSFM, METTL21B, METTL1, CYP27B1, MARCH9, CDK4, AGAP2, OS9 | Nivel de inhibina QTL (71509, 71314, 71315, 71316, 71317, 71358, 71359, 71319, 71407, 71320); Color del pelaje QTL (37323, 37324, 37325) |
| 5 | 40 | 62920850 | 66830677 | ACTR6, NR1H4, ANO4, SLC5A8, UTP20, PARPBP, NUP37, GNPTAB | Grosor de la grasa en la 12a costilla QTL (20283); Facilidad para parir QTL (126849); Mortinatos QTL (126850); Duración de la gestación QTL (15409, 15410, 15411, 15412); Perfil de ácidos grasos en la leche C14 QTL (34847); Rendimiento de carne magra QTL (36911) |
| 5 | 14 | 103006312 | 104069660 | WC1-12, WC1.3, CLSTN3 | Fuerza de corte QTL (121704) |
| 6 | 19 | 24733464 | 26517604 | PPP3CA, EMCN, DNAJB14 | Mastitis clínica QTL (25244); Peso corporal (al nacer) QTL (67220, 67402, 66543); Aumento de peso corporal QTL (67403, 67639); Facilidad del parto QTL (106434) |
| 6 | 16 | 73543373 | 75270817 | KIAA1211, AASDH, SRP72, NOA1, IGFBP7 | Firmeza de la cuajada QTL (95977); Pigmentación del área ocular QTL (37389); Peso corporal (al nacer) QTL (66358); Peso corporal (al destete) QTL (67229); Peso corporal (añal) QTL (67230); Aumento de peso corporal QTL (67231); Rendimiento de leche (EBV) QTL (16233); Rendimiento de grasa de la leche QTL (16234); Porcentaje de grasa en la leche (EBV) QTL (16235); Porcentaje de proteína en la leche QTL (16236); Tasa de no retorno (EBV) QTL (16237); Intervalo de la primera inseminación a la última QTL (16238); Rendimiento de proteína de la leche (desviación de las hijas) QTL (26193, 26196); Rendimiento de grasa de la leche (desviación de las hijas) QTL (25834); Rendimiento de leche (desviación de las hijas) QTL (25421); Porcentaje de kappa-caseína en la leche QTL (111104, 112361) |
| 7 | 9 | 4358132 | 4953801 | CRTC1, CRLF1, ELL, SSBP4, PGPEP1 | Aumento de peso corporal QTL (67930) |
| 7 | 56 | 94824183 | 100624993 | NR2F1, FAM172A, KIAA0825, SLF1, MCTP1, FAM81B, TTC37, PCSK1, ERAP1, LNPEP, LIX1 | Contenido de zinc QTL (24065); Grosor de la grasa en la 12a costilla QTL (24649, 24650); Fuerza de corte QTL (20767, 31580, 37955); Contenido de hierro QTL (23257, 23258); Rendimiento de carne magra QTL (37087); Peso en canal QTL (122454); Puntaje de suavidad QTL (36406) |
| 8 | 45 | 46925080 | 55060853 | SMC5, TRPM3, ABHD17B, C8H9orf85, GDA, ZFAND5, TMC1, RFK, PRUNE2, GNA14, VPS13A, GNAQ, CEP78 | Aumento diario promedio QTL (20937); Contenido de zinc QTL (24066); Rendimiento de leche QTL (121783); Rendimiento de grasa en la leche QTL (121784); Rendimiento de proteína en la leche QTL (121785); Porcentaje de proteína en la leche QTL (121786) |
| 8 | 29 | 74227581 | 77959797 | DOCK5, CDCA2, PPP2R2A, PTK2B, GULO, B4GALT1, NFX1, UBE2R2, KIF24, DNAI1, CCL19 | Rendimiento de proteína en la leche (desviación de las hijas) QTL (26264, 26268); Rendimiento de grasa en la leche (desviación de las hijas) QTL (25878); Rendimiento de leche (desviación de las hijas) QTL (25466); Grosor de la grasa en la 12a costilla QTL (122442); Intervalo al primer estro después del parto QTL (29993, 30322) |
| 10 | 12 | 28781938 | 29967808 | FMN1, AVEN, RYR3 | Contenido de ácido docosapentaenoico QTL (31774); Contenido de ácido graso insaturado omega-3 QTL (31780); Facilidad de parto (materna) QTL (44416); Tasa de embarazo de las hijas QTL (44417); Mortinatos (maternos) QTL (44418); ángulo de la pata QTL (44419); Conformación de las patas y las piernas QTL (44420); Tipo de PTA QTL (44421); Ubicación de los pezones (al frente) QTL (44422); Inserción de la ubre QTL (44423); Mérito neto QTL (44424); Longitud de la vida productiva QTL (44425); Posición de las patas traseras - vista posterior QTL (44426); Posición de las patas traseras - vista posterior QTL (44427); Altura de la ubre QTL (44428); Ancho de grupa QTL (44429); Facilidad para parir QTL (44430); Puntaje de células somáticas QTL (44431); Mortinatos QTL (44432); Estatura QTL (44433); Profundidad de la ubre QTL (44434) |
| 10 | 52 | 30065944 | 39409071 | DPH6, C10H15orf41, MEIS2, RTF1, MGA, MAPKBP1, SPTBN5, PLA2G4E, VPS39, CAPN3, STARD9, CDAN1, UBR, SCG5, AQR | Ingesta de pienso residual QTL (23793); Contenido de hierro QTL (24060); Facilidad para parir QTL (15185, 44566); Ingesta de material seca QTL (140483); Profundidad corporal QTL (44557, 44572, 44586, 44672); Forma láctea QTL (44558, 44573, 44587, 44673); Tasa de embarazo de las hijas QTL (44559); Tipo de PTA QTL (44560, 44575, 44589, 44675); Mérito neto QTL (44561); Duración de la vida productiva QTL (44562); Posición de las patas traseras - vista posterior QTL (44563, 44578, 44592, 44677); Posición de los pezones - trasera QTL (44564, 44579, 44593); Altura de la ubre QTL (44565, 44580, 44594, 44678); Puntaje de células somáticas QTL (44567); Mortinatos QTL (44568); Estatura QTL (44569, 44582, 44596, 44680); Longitud de los pezones QTL (44570); Hendidura de la ubre QTL (44571, 44584, 44598); Tasa de concepciones QTL (107126, 107124); Conformación de las patas y las piernas QTL (44574, 44588, 44674); Posición de los pezones - frontal QTL (44576, 44590); Inserción de la ubre QTL (44577, 44591, 44676); Ancho de grupa QTL (44581, 44595, 44679); Fortaleza QTL (44583, 44597, 44681); Profundidad de la ubre QTL (44585, 44599); Fuerza de corte QTL (37956, 20778) |
| 10 | 60 | 42713194 | 49942490 | KLHDC2, NEMF, SOS2, CDKL1, MAP4K5, TRIM9, CSNK1G1, FAM96A, DAPK2, HERC1, CA12, APH1B, TLN2, VPS13C, FRMD6, GNG2, NID2, PTGDR, ZNF609, TRIP4, RORA, ICE2 | Forma láctea QTL (44720); Tasa de embarazo de las hijas QTL (44721); Mérito neto QTL (44722); Duración de la vida productiva QTL (44723); Porcentaje de proteína en la leche QTL (44724, 105566); Facilidad para parir QTL (44725); Puntaje de células somáticas QTL (44726); Mortinatos QTL (44727); Porcentaje de kappa-caseína glicosilada en la leche QTL (116745, 111446); Rendimiento de la grasa en la leche (desviación de las hijas) QTL (25900); Resistencia a las garrapatas QTL (135843); Ingesta de materia seca QTL (131016, 131002); Peso corporal (al nacer) QTL (68188); Aumento del peso corporal QTL (68135); Contenido de ácido eicosapentaenoico QTL (31775); Contenido de ácido graso insaturado omega-3 QTL (31781) |
| 10 | 81 | 50086640 | 59882200 | DMXL2, GLDN, TNFAIP8L3, SPPL2A, TRPM7, MYO1E, CCNB2, ADAM10, LIPC, ALDH1A2, POLR2M, MYZAP, CGNL1, TCF12, NEDD4, PRTG, RAB27A, UNC13C, WDR72, FAM214A, MYO5A, GNB5, MAPK6 | Resistencia a las garrapatas QTL (135792); Peso corporal (al destete) QTL (23796); Puntuación de la ubre, QTL de turgencia de la ubre (106594); Intervalo al primer estro después del parto QTL (28678, 28652); Grosor de la grasa en la 12a costilla QTL (122444); Susceptibilidad bovina a las enfermedades respiratorias QTL (95662); Peso corporal (al destete) QTL (23795); Tasa de concepciones QTL (138570); Contenido de ácido graso C22:1 QTL (20512); Facilidad para parir QTL (30516); Fuerza de corte QTL (106393) |
| 10 | 63 | 60004650 | 66740333 | SHC4, CEP152, FBN1, SLC12A1, MYEF2, SEMA6D, FERMT2, DDHD1, GABPB1, ATP8B4, FAM227B, GALK2 | Facilidad para parir QTL (30516); Fuerza de corte QTL (106393); Índice de partos QTL (30512); Tasa de embarazos QTL (65946); Contenido de zinc en la leche QTL (70034); Tasa de concepciones QTL (107130); Profundidad corporal QTL (44827, 44850, 44863); Forma láctea QTL (44828, 44851, 44864); Tipo de PTA QTL (44829, 44853, 44865); Inserción de la ubre QTL (44830, 44855, 44867); Altura de la ubre QTL (44831, 44857, 44869); Ancho de grupa QTL (44832, 44858, 44870); Estatura QTL (44833, 44859, 44872); Fortaleza QTL (44834, 44860, 44873); Hendidura de la ubre QTL (44835, 44861, 44874); Profundidad de la ubre QTL (44836, 44862, 44875); Conformación de las patas y las piernas (44852); Posición de los pezones - frontal QTL (44854, 44866); Posición de los pezones - posterior QTL (44856, 44868); Aumento de peso corporal QTL (68136), Puntaje de células somáticas QTL (44871); Peso corporal (al destete) QTL (106643); Duración de la gestación QTL (15473, 15474) |
| 10 | 3 | 98223275 | 98398966 | -- | Peso corporal (al destete) QTL (24729) |
| 10 | 14 | 100918254 | 101806982 | GALC, KCNK10, ZC3H14, EML5 | Área del músculo Longissimus QTL (138414) |
| 11 | 28 | 2254541 | 4099982 | ASTL, CIAO1, KANSL3, LMAN2L, CNNM3, SEMA4C, FAM178B | Porcentaje de kappa-caseína glicosilada QTL (116678) |
| 11 | 31 | 36863940 | 40183335 | ZAP70, TMEM131, VWA3B, CNGA3, COA5, MGAT4A, KIAA1211L, ACYP2, SPTBN1, EML6, RTN4, CCDC88A, PPP4R3B, CCDC85A | Contenido de riboflavina en la leche QTL (64136); Profundidad corporal QTL (45332); Facilidad para parir (materna) QTL (45333); Ángulo de la pata QTL (45334); Conformación de las patas y las piernas QTL (45335); Porcentaje de grasa en la leche QTL (45336); Tipo de PTA QTL (45337); Inserción de la ubre QTL (45338); Rendimiento de grasa de la leche QTL (45339); Rendimiento de la leche QTL (45340); Mérito neto QTL (45341); Porcentaje de proteína en la leche QTL (45342); Rendimiento de proteína de la leche (45343); Posición de las patas traseras - vista posterior QTL (45344); Altura de la ubre QTL (45345); Ancho de grupa QTL (45346); Facilidad para parir QTL (45347); Mortinatos QTL (45348); Estatura QTL (45349); Fortaleza QTL (45350); Profundidad de la ubre QTL (45351); Fuerza de corte QTL (36420, 36421); Turgencia de la ubre (106598); Ingesta de materia seca QTL (121918); Intervalo entre partos QTL (121653, 121654) |
| 13 | 25 | 61014137 | 63715266 | ANGPT4, TBC1D20, HM13, TTLL9, CCM2L, NOL4L, DNMT3B, SUN5, BPIFB4, SNTA1 | Longitud de los pezones QTL (47334); Intervalo al primer estro después del parto QTL (28680); Contenido de hierro en la leche QTL (70225); Circunferencia del escroto QTL (119789) |
| 14 | 10 | 4202927 | 4797465 | KCNK9, TRAPPC9, AGO2 | Porcentaje de grasa de la leche QTL (33087, 35538, 47482, 61978, 32849, 35540, 14973, 33665, 33581, 35542, 33227, 32960); Porcentaje de proteína en la leche QTL (35774, 113879, 47486, 35776, 113990); Rendimiento de proteína de la leche QTL (35306, 35308, 14927); Rendimiento de leche QTL (35423, 35425, 14900); Porcentaje de caseína en la leche QTL (108945); Contenido de riboflavina en la leche QTL (64384); Puntaje de células somáticas QTL (64598); Ángulo de la pata QTL (47480); Conformación de las patas y piernas QTL (47481); Rendimiento de grasa de la leche QTL (47483, 35359, 35360, 31117, 35362); Mérito neto QTL (47484); Duración de la vida productiva QTL (47485); Posición de las patas traseras - vista posterior QTL (47487); Posición de las patas traseras - vista lateral QTL (47488); Facilidad para parir QTL (47489); Mortinatos QTL (47490); Estatura QTL (47491); Fortaleza QTL (47492); Porcentaje de ácido oleico en la leche QTL (32556) |
| 14 | 18 | 33747933 | 35216192 | C14H8orf34, PREX2, CPA6 | Puntuación de la ubre, Turgencia de la ubre QTL (106736); Edad al primer parto QTL (140104); Circunferencia del escroto QTL (138445) |
| 16 | 25 | 2020646 | 3805373 | PIK3C2B, NFASC, CNTN2, DSTYK, TMCC2, MFSD4A, NUCKS1, PM20D1, CTSE, SRGAP2 | Profundidad corporal QTL (48079); Facilidad para parir (maternal) QTL (48080); Forma láctea QTL (48081); Tasa de embarazo de las hijas QTL (48082); Tipo de PTA QTL (48083); Duración de la vida productiva QTL (48084); Facilidad para parir QTL (48085); Longitud de los pezones QTL (48086); Hendidura de la ubre QTL (48087); Porcentaje de ácido mirístico en la leche QTL (56623); Aumento de peso corporal QTL (68840) |
| 16 | 11 | 5715970 | 7838108 | KCNT2 | Porcentaje de grasa de la leche QTL (34602) |
| 16 | 73 | 25709923 | 26613598 | -- | -- |
| 20 | 40 | 21402231 | 22154025 | GPBP1 | Aumento de peso corporal QTL (69057) |
| 20 | 36 | 27105033 | 30747366 | HCN1, EMB, PARP8 | Facilidad para parir QTL (30553); Aumento promedio diario QTL (131122); Duración de la vida productiva QTL (123122, 123133); Porcentaje de proteína de la leche QTL (105307, 105349, 105182, 105395, 105073); Porcentaje de proteína de la leche (desviación de las hijas) QTL (26866, 26984); Rendimiento de la leche (desviación de las hijas) QTL (25661); Peso corporal QTL (65983) |
| 20 | 15 | 45232289 | 47209012 | CDH9 | -- |
| 21 | 6 | 6388853 | 6900116 | CERS3, ADAMTS17 | Profundidad corporal QTL (50966); Facilidad para parir (materna) QTL (50967); Tasa de embarazo de las hijas QTL (50968); Mortinatos (materno) QTL (50969); Ángulo de la pata QTL (50970); Conformación de patas y piernas QTL (50971); Tipo de PTA QTL (50972); Posición de los pezones - frontal QTL (50973); Inserción de la ubre QTL (50974); Mérito neto QTL (50975); Duración de la vida productiva QTL (50976); Porcentaje de proteína de la leche QTL (50977); Altura de la ubre QTL (50978); Ancho de grupa QTL (50979); Facilidad para parir QTL (50980); Estatura QTL (50981); Fortaleza QTL (50982); Profundidad de la ubre QTL (50983) |
| 21 | 27 | 7068248 | 9209367 | MEF2A, LRRC28, TTC23, SYNM, IGF1R, PGPEP1L | Edad de la pubertad QTL (21135, 21136); Tasa de concepción del progenitor QTL (124003); Área del músculo Longissimus QTL (22856); Ingesta de material seca QTL (23894); peso corporal QTL (22618); Intervalo desde la primera inseminación a la última QTL (138530); Inseminaciones por concepción QTL (138531) |
| 21 | 9 | 63970044 | 65102247 | -- | -- |
| 22 | 18 | 21888915 | 23828165 | TPR1, SUMF1, CNTN4 | Susceptibilidad a M. paratuberculosis QTL (127097); Peso corporal (al nacer) QTL (23910) |
| 22 | 1 | 50772465 | 50772465 | -- | -- |
| 24 | 6 | 33291018 | 33669073 | LAMA3, ANKRD29, NPC1, TMEM241 | Rendimiento de proteína de la leche QTL (123993) |
| 29 | 7 | 50202589 | 50336324 | TSPAN32 | Aumento diario promedio QTL (102011, 102012); Índice de crecimiento QTL (102013) |
Discusión
Análisis de componentes principales
El ACP es una herramienta útil que permite a los ganaderos identificar a los individuos genéticamente diferentes dentro de su hato con base en los genotipos. De hecho, la representación gráfica de los individuos es una herramienta inmediata fácil de interpretar que no requiere de ninguna destreza específica por parte de los ganaderos, salvo el concepto de que los puntos cercanos son más similares, y los distantes, diferentes. Es posible identificar a las hembras bien diferenciadas en este hato, sobre todo por su progenitor, si bien existe un grupo que parece ser genéticamente similar pero que procede de progenitores diversos. Los sementales progenitores 1 y 5 son responsables de los grupos marginales que vemos, y sus derivaciones son Canadá e Italia. Por lo tanto, como lo propuso el ganadero que participó en este estudio, esta ilustración es benéfica para determinar el análisis a posteriori de sus estrategias de apareamiento y si los sementales que eligió están contribuyendo a sus intentos de limitar la reducción de la variabilidad genética del hato. El ACP también es útil para la selección de los sementales, puesto que el ganadero puede verificar si los sementales provenientes de determinados países influyen o no en la distribución genética en la población. Asimismo, es posible visualizar cómo los valores genómicos, en este caso de GPFT, coinciden con diferentes individuos. Los GPFT de valor similar no se agruparon, lo que indica que ni el progenitor ni su origen influyeron en los GPFT de las hembras de este hato. Por ejemplo, el semental 1 aportó a los animales valores de valores de GPFT de entre 1,500 y 3,000, y el semental 5, valores de entre 1,000 y 2,500.
Coeficientes de consanguinidad
El resultado obtenido aquí concuerda con otro estudio realizado en ganado Holstein italiano, donde el F PED fue de 0.044 y el F ROH fue significativamente diferente de 0, con un r igual a 0.453 (P<0.01). La práctica de una selección artificial intensa y precisa a lo largo de muchos años ha dado como resultado índices elevados de ganancia genética; sin embargo, las altas tasas de ganancia se han visto acompañadas de un gran aumento de la endogamia17. Además, a medida que se reducen los tamaños de la población, la probabilidad de aparearse entre parientes consanguíneos aumenta, sobre todo en hatos pequeños o razas locales13. Los productores han dependido en gran medida de los pedigríes para calcular el coeficiente de consanguinidad dentro del hato, pero esta cifra sólo puede crecer con cada generación. La interconexión entre pedigríes da una proporción esperada, no real, de la identidad genómica por ascendencia entre individuos, y se anticipa que las estimaciones basadas en genotipos proporcionan una mayor exactitud respecto a dicha interconexión18. En la práctica, la información del pedigrí es difícil de obtener, puede no ser confiable, y rara vez es evaluada en cuanto a la consanguinidad surgida de antepasados comunes19. Cuando examinamos los coeficientes basados en pedigrí de este hato, consideramos que los individuos procedían de hasta diez retrocruzamientos. Sin embargo, en ausencia de datos del pedigrí, se puede utilizar la extensión de un genoma bajo ROH para inferir aspectos de la historia reciente de la población, incluso a partir de relativamente pocas muestras3,9. McQuillan et al9 revelaron que el F ROH esa fuertemente correlacionado con el coeficiente de consanguinidad estimado a partir del pedigrí (e= 0.86), y se ha visto que los valores del F ROH son preferibles al F PED, puesto que se los considera un reflejo más realista del nivel de consanguinidad15,19. Sin embargo, es importante señalar que el establecimiento de los parámetros utilizados para derivar los ROH es esencial para explicar correctamente los efectos de la densidad de los SNP20. En este conjunto de datos percibimos una relación lineal entre el F ROH y el F PED. El coeficiente de correlación de Pearson proporciona una correlación positiva con un r= 0.453. Nuestra correlación concuerda con la de otros estudios que utilizan ganado genotipificado con una densidad similar15,20 pero más baja que la de otros ganados genotipificados a una densidad media o alta3,5. Es posible, entonces, que el coeficiente de correlación se vea parcialmente afectado por el pequeño número de animales en este estudio, así como por el uso de un chip SNP de baja densidad. Además, el F ROH fue mayor que el F PED promedio en 35 animales, lo que implica que los coeficientes de consanguinidad pasados en pedigrí podrían ser subestimados.
Los valores bajos generales indican un nivel bajo de consanguinidad en la población y un número relativamente alto de individuos en un estado heterocigoto. Esto puede estar relacionado con el resultado de la estrategia de apareamiento específica implementada por el responsable del hato de la Universidad, coautor de este trabajo, puesto que él señaló que su meta era incrementar la variabilidad genética y mantener un nivel de endogamia bajo. Por ello, esta información genética está proveyendo una retroalimentación única sobre los esfuerzos exitosos para mantener el nivel de endogamia dentro de esta población. Las razas lecheras en particular han sido objeto de una selección más intensiva y pueden estar relacionadas con el reciente aumento de apareamientos endogámicos que resulta del uso de un número pequeño de sementales con altos méritos genéticos para la inseminación artificial5. Por este motivo es importante entender la consanguinidad que se da al nivel genético, así como determinar si la variabilidad genética dentro del propio hato está siendo bien mantenida o no.
Tramos de homocigosidad y genes asociados
Los QTL más comúnmente observados dentro de estas regiones reportadas en la Tabla 2 incluyeron el porcentaje de proteína en la leche, el porcentaje de grasa de la leche, la facilidad para parir, el tipo de PTA y la estatura. Después de alinear los SNP con la referencia BTA U 5.0.1 se identificaron 260 genes anotados como consistentes con el primer 5% de los SNP, mientras que 37 genes permanecieron en el primer 1%. Los genes del primer 1% residían en los cromosomas 5 y 10; no se identificaron genes anotados con SNP en BTA 16. Posteriormente, STRING identificó redes entre estos genes, de los cuales 142 formaban parte de alguna especie de red (Figura 5). A éstas se les practicó también una ontología de genes en PANTHER. A los genes dentro del primer 5% correspondieron a 12 procesos biológicos (Figura 6) y a 70 vías (no se muestran datos). La vía que incluyó el mayor número de genes (n= 12) fue la vía del receptor de la hormona liberadora de gonadotropina, mientras que otras vías que compartían genes eran la vía de señalización mediada por el receptor tipo 5HT2, la vía de señalización de endotelina, la vía de señalización de inflamación mediada por quimoquina y citoquina, la vía de señalización de integrina y la vía de señalización del sitio de integración de tipo sin alas (WNT). Los procesos biológicos respectivo que más comúnmente se compartieron entre genes (n= 16) fueron “procesos celulares”, específicamente la comunicación entre células, mientras que otra porción grande (n= 79) desempeña un papel en los procesos metabólicos (Figura 6).

Figura 5 Red de genes que corresponden con SNP en el primer 5% de incidencia de tramos comunes obtenida de la base de datos en línea STRING
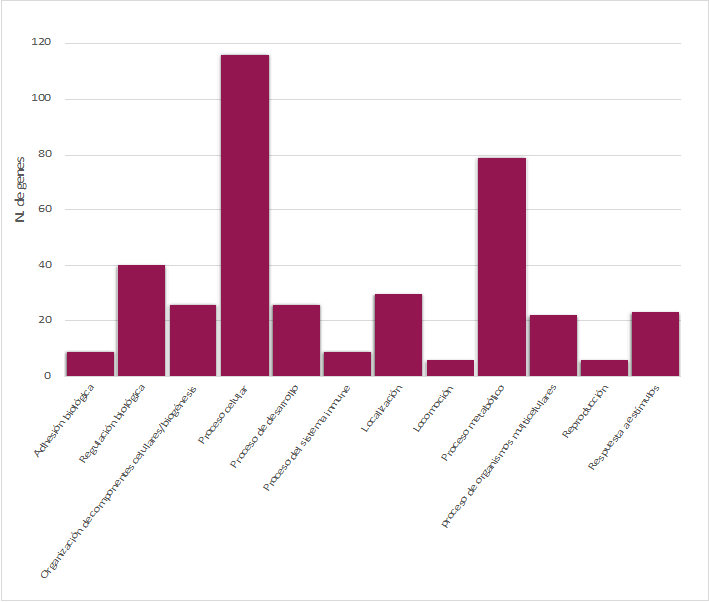
Figura 6 Proceso biológico de genes correspondiente al 5% del top de incidencia de corridas comunes con SNP
Las prácticas de selección intensa de sementales, de la inseminación artificial y de la transferencia de embriones han ocupado un lugar muy destacado en algunas razas, reduciendo los tamaños efectivos de las poblaciones y la diversidad genética y afectando los niveles de homocigosidad3. Los efectos nocivos asociados con el incremento de la homocigosidad que surge de la consanguinidad tienen una predisposición a reducir las ganancias genéticas, lo que implica una clara pérdida de la variabilidad genética15. Los ROH permiten comprender mejor esta variabilidad, y son segmentos homocigotos continuos que resultan comunes tanto en los individuos como en las poblaciones. La capacidad de estos segmentos de darnos un atisbo de los eventos genéticos de una población los convierte en una herramienta útil que proporciona información sobre la evolución de la población a través del tiempo, permitiendo a los productores mantener la diversidad y la idoneidad en su raza de ganado21.
Además, los ROH brindan información útil sobre las relaciones genéticas entre individuos, ayudando a minimizar el índice de consanguinidad y a exponer las variables nocivas presentes en el genoma21. Los ROH largas surgen como resultado de una endogamia reciente, mientras que los ROH más cortos pueden indicar efectos ancestrales más distantes, tales como los efectos fundadores de la raza3. De hecho, la presencia de segmentos de más de 10 Mb de longitud es trazable a la consanguinidad con antepasados comunes recientes de hace sólo cinco generaciones22, y el 66 % de los animales comprendidos en este estudio presentaron por lo menos un ROH de más de 10 Mb. Esto significa que la mayoría de los individuos de esta población se derivaron de antepasados comunes recientes y son el producto de una endogamia reciente, en conformidad con lo que vemos por sus coeficientes de consanguinidad.
También se identificaron los ROH como un medio de facilitar la comprensión de las regiones genéticas conservadas en el seno del hato. Los ROH de menos de 5 Mb se caracterizaron recientemente como cortos23. Estos ROH cortos se pueden relacionar con un efecto de selección positiva ancestral más distante debido a eventos de recombinación generados por la ruptura de los cromosomas largos en segmentos por meiosis repetidas que, por ende, reduce su tamaño13,24. La menor densidad de los SNP, como la que se utilizó en este estudio, tiende a inflar los ROH3. Dado que vemos principalmente ROH cortos en este hato (ROH promedio=4.66 Mb), incluso con la inflación potencial, podemos decir con confianza que dentro de esta población se ha mantenido bien el nivel de consanguinidad, el cual es bajo y reciente, a juzgar por la presencia de ROH largos en la mayoría de los individuos y, a la vez, de ROH cortos en general en el hato. Este resultado se identifica con otro estudio que estableció que el ganado Holstein italiano muestra un número elevado de segmentos de ROH relacionados con una consanguinidad antigua13.
Se encontró que los genes que se localizan en las regiones homocigotas extendidas correspondientes a los SNP en el primer 5% de los tramos comunes intervienen en gran medida en los procesos de supervivencia. Esto indica que en los esfuerzos de selección se han mantenido los procesos biológicos básicos, como la comunicación y el metabolismo celulares. Sin embargo, los QTL dentro de estas regiones genéticas demuestran que éstas coinciden con facetas benéficas de la producción tales como el porcentaje de proteína en la leche, el porcentaje de grasa de la leche y la facilidad para parir, aunque es posible que éstas sean específicas de la raza Holstein-Friesian debido a rigurosos esfuerzos de selección a lo largo del tiempo. Además, los genes del primer 1% (BTA 5, 10 y 16) también se correlacionaron con los procesos de supervivencia y de desarrollo. Entre estos procesos se incluían la proliferación de las células (GNG2, ADAM10, ALDH1A2), la función metabólica (SCG5, PTGDR, RORA, MYO1E, LIPC, ALDH1A2, MYO5A, GALK2), el desarrollo de los órganos (ANKS1B, MYO1E, CCNB2, ALDH1A2, PRTG, MAPK6) y la inmunidad (NEDD4). No se identificaron genes en BTA 16 debido a una mala anotación; pero varios genes en los cromosomas BTA 5 y 10 han sido bien caracterizados en diferentes fenotipos en el ganado. La proteína de unión al ADN PARP1 (también conocida como PARPBP) en los BTA 5 y 10 ha sido asociada con el porcentaje de grasa en la tercera etapa de la lactancia en el ganado Jersey, mientras que la repetición de anquirina y el dominio con motivo alfa estéril 1B (ANKS1B), también en BTA5, ha sido asociado con la incidencia de encefalopatía espongiforme bovina (EEB) en familias de ganado25,26. En tanto que se ha visto que los genes en el cromosoma BTA 10 intervienen en gran medida en el metabolismo de la grasa y la ingesta de pienso. Por ejemplo, se ha vinculado la secretogranina V y la subunidad gamma 2 de la proteína G (GNG2) con la regulación de los procesos del metabolismo hormonal que intervienen en la ingesta de pienso en el ganado Holstein27,28, mientras que se encontró que el factor de transcripción 12 (TCF12) está asociado a los rasgos lípidos y organolépticos en las razas europeas Bos taurus29. También se encontró la subunidad GNG2 en una región asociada con la susceptibilidad a la infección por Mycobacterium avium subespecie Paratuberculosis (Map)30. Por último, se identificó que el protema 6 que contiene dominio FERM 6 (FRMD6) se ubica en una región de selección positiva en la raza N’Dama31. Las regiones en los cromosomas BTA 10 y 16 que albergan el principal 1% de los SNP también coincidieron con otros estudios. Los estudios de ganado Holstein fueron particularmente similares, con una selección positiva al nivel de los cromosomas BTA 10 en 50-560 Mb y un QTL putativo identificado en 34.7-56.9 en el cromosoma BTA 10 para la cuenta de células somáticas y la tasa de no retorno a 90 días (efecto paterno)20,32,33. Otro estudio identificó una elevada proporción de ROH en BTA 16, mientras que otro más encontró selección en las regiones 24.7-26.7 y 26.5-28.5 en BTA 163,20.
Conclusiones e implicaciones
Valiéndose de una población de 44 cabezas de ganado italiano Holstein-Friesian de la granja experimental de la Universidad y de paneles de SNP de baja densidad para la genotipificación, se estimaron varios aspectos de la variabilidad genética dentro de la población del hato incluyendo los coeficientes de consanguinidad y los tramos de homocigosidad. El enfoque proporcionó exitosamente una herramienta para monitorear la eficacia de las estrategias de apareamiento operadas en la población de la granja y conocimientos para la gestión de la endogamia. Los genotipos SNP pueden proporcionar eficazmente los coeficientes de consanguinidad en ausencia de un pedigrí, mientras que los tramos de homocigosidad pueden proveer información adicional sobre las regiones genéticas bajo selección positiva, e indicar así el carácter evolutivo de la población. Si los ganaderos genotipifican más ampliamente a sus hembras jóvenes, podrán construir una base de datos con la información correspondiente y, como resultado, tomar decisiones relativas a la selección y manejo del hato. Cabe señalar que la gestión de la endogamia es extremadamente importante para reducir la incidencia de la enfermedad recesiva mendeliana y que el enfoque genético está proporcionando una posibilidad innovadora y precisa. En general los ganaderos genotipifican a hembras con un chip SNP de baja densidad, que ha demostrado ser una herramienta valiosa para fines de gestión genética. La difusión de este enfoque a los ganaderos puede dar lugar a una genotipificación rutinaria de las hembras jóvenes y proporcionarles así información para lograr un amplio impacto en la gestión genética de la endogamia.
Agradecimientos
Este trabajo se realizó en parte con el apoyo del Instituto Nacional de Alimentos y Agricultura del Departamento de Agricultura de Estados Unidos (USDA), con la beca complemento 2017-38420-26779. El resto del apoyo se recibió a través de fondos internos de Azienda Agraria Didattico Sperimentale Angelo Menozzi.
REFERENCIAS
1. Toro MA, Meuwissen TH, Fernandez J, Shaat I, Maki-Tanila A. Assessing the genetic diversity in small farm animal populations. Animal 2011;5(11):1669-83. [ Links ]
2. Ouborg NJ, Pertoldi C, Loeschcke V, Bijlsma RK, Hedrick PW. Conservation genetics in transition to conservation genomics. Trends Genet 2010;26(4):177-87. [ Links ]
3. Purfield DC, Berry DP, McParland S, Bradley DG. Runs of homozygosity and population history in cattle. BMC Genet 2012;13:70. [ Links ]
4. Ferencakovic M, Hamzic E, Gredler B, Solberg TR, Klemetsdal G, Curik I, et al. Estimates of autozygosity derived from runs of homozygosity: empirical evidence from selected cattle populations. J Anim Breed Genet 2013;130(4):286-93. [ Links ]
5. Marras G, Gaspa G, Sorbolini S, Dimauro C, Ajmone-Marsan P, Valentini A, et al. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim Genet 2015;46(2):110-21. [ Links ]
6. Schwarzenbacher H. Analysis of genome regions showing strong inbreeding in Brown Swiss and Fleckvieh cattle. Interbull Bulletin 2011;44:130-133. [ Links ]
7. Schefers JM, Weigel KA. Genomic selection in dairy cattle: Integration of DNA testing into breeding programs. Animal Frontiers 2012;2(1):4-9. [ Links ]
8. Pryce JE, Hayes BJ, Goddard ME. Genotyping dairy females can improve the reliability of genomic selection for young bulls and heifers and provide farmers with new management tools. In: Proc 38th ICAR Session. Cork 2012. https://www.icar.org/index.php/icar-meetings-news/38th-session-cork-ireland/. [ Links ]
9. McQuillan R, Leutenegger AL, Abdel-Rahman R, Franklin CS, Pericic M, Barac-Lauc L, et al. Runs of homozygosity in European populations. Am J Hum Genet 2008;83(3):359-372. [ Links ]
10. Kinghorn B. Pedigree Viewer. Pedigree Viewer. 6.5f ed.; 2015. https://bkinghor.une.edu.au/pedigree.htm [ Links ]
11. Curik I, Ferencakovic M, Solkner J. Inbreeding and runs of homozygosity: A possible solution to an old problem. Livest Sci 2014;166:26-34. [ Links ]
12. Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 2017;45(D1):D362-D368. [ Links ]
13. Mastrangelo S, Tolone M, Di Gerlando R, Fontanesi L, Sardina MT, Portolano B. Genomic inbreeding estimation in small populations: evaluation of runs of homozygosity in three local dairy cattle breeds. Animal 2016;10(5):746-54. [ Links ]
14. Kim ES, Cole JB, Huson H, Wiggans GR, Van Tassell CP, Crooker BA, et al. Effect of artificial selection on runs of homozygosity in U.S. Holstein cattle. PLoS One 2013;8(11):e80813. [ Links ]
15. Peripolli E, Stafuzza NB, Munari DP, Lima ALF, Irgang R, Machado MA, et al. Assessment of runs of homozygosity islands and estimates of genomic inbreeding in Gyr (Bos indicus) dairy cattle. BMC Genomics 2018;19(1):34. [ Links ]
16. Hu ZL, Park CA, Reecy JM. Developmental progress and current status of the Animal QTLdb. Nucleic Acids Res 2016;44(D1):D827-33. [ Links ]
17. Rodriguez-Ramilo ST, Fernandez J, Toro MA, Hernandez D, Villanueva B. Genome-wide estimates of coancestry, inbreeding and effective population size in the Spanish Holstein population. PLoS One 2015;10(4):e0124157. [ Links ]
18. Visscher PM, Medland SE, Ferreira MA, Morley KI, Zhu G, Cornes BK, et al. Assumption-free estimation of heritability from genome-wide identity-by-descent sharing between full siblings. Plos Genet 2006;2(3):e41. [ Links ]
19. Keller MC, Visscher PM, Goddard ME. Quantification of inbreeding due to distant ancestors and its detection using dense single nucleotide polymorphism data. Genetics 2011;189(1):237-249. [ Links ]
20. Signer-Hasler H, Burren A, Neuditschko M, Frischknecht M, Garrick D, Stricker C, et al. Population structure and genomic inbreeding in nine Swiss dairy cattle populations. Genet Sel Evol 2017;49(1):83. [ Links ]
21. Peripolli E, Munari DP, Silva M, Lima ALF, Irgang R, Baldi F. Runs of homozygosity: current knowledge and applications in livestock. Anim Genet 2017;48(3):255-271. [ Links ]
22. Howrigan DP, Simonson MA, Keller MC. Detecting autozygosity through runs of homozygosity: a comparison of three autozygosity detection algorithms. BMC Genomics 2011;12:460. [ Links ]
23. Mastrangelo S, Ciani E, Sardina MT, Sottile G, Pilla F, Portolano B, et al. Runs of homozygosity reveal genome-wide autozygosity in Italian sheep breeds. Anim Genet 2018;49(1):71-81. [ Links ]
24. Kirin M, McQuillan R, Franklin CS, Campbell H, McKeigue PM, Wilson JF. Genomic runs of homozygosity record population history and consanguinity. PLoS One 2010;5(11):e13996. [ Links ]
25. Murdoch BM, Clawson ML, Laegreid WW, Stothard P, Settles M, McKay S, et al. A 2cM genome-wide scan of European Holstein cattle affected by classical BSE. BMC Genet 2010;11:20. [ Links ]
26. Oliveira HRd, Silva FF, Brito LF, Jamrozik J, Lourenco DAL, Schenkel FS. Genome-wide association study for milk, fat and protein yields in different lactation stages in Canadian Holstein and Jersey cattle. World Congress on Genetics Applied to Livestock Production, Auckland, New Zealand. 2018. [ Links ]
27. Xi YM, Yang Z, Wu F, Han ZY, Wang GL. Gene expression profiling of hormonal regulation related to the residual feed intake of Holstein cattle. Biochem Biophys Res Commun 2015;465(1):19-25. [ Links ]
28. Salleh MS, Mazzoni G, Hoglund JK, Olijhoek DW, Lund P, Lovendahl P, et al. RNA-Seq transcriptomics and pathway analyses reveal potential regulatory genes and molecular mechanisms in high- and low-residual feed intake in Nordic dairy cattle. BMC Genomics 2017;18(1):258. [ Links ]
29. Dunner S, Sevane N, Garcia D, Leveziel H, Williams JL, Mangin B, et al. Genes involved in muscle lipid composition in 15 European Bos taurus breeds. Anim Genet 2013;44(5):493-501. [ Links ]
30. Kiser JN, Neupane M, White SN, Neibergs HL. Identification of genes associated with susceptibility to Mycobacterium avium ssp. Paratub erculosis (Map) tissue infection in Holstein cattle using gene set enrichment analysis-SNP. Mamm Genome 2017;1-11. doi: 10.1007/s00335-017-9725-4. [ Links ]
31. Taye M, Lee W, Jeon S, Yoon J, Dessie T, Hanotte O, et al. Exploring evidence of positive selection signatures in cattle breeds selected for different traits. Mamm Genome 2017;28(11-12):528-541. [ Links ]
32. Kuhn C, Bennewitz J, Reinsch N, Xu N, Thomsen H, Looft C, et al. Quantitative trait loci mapping of functional traits in the German Holstein cattle population. J Dairy Sci 2003;86(1):360-368. [ Links ]
33. Bovine HapMap C, Gibbs RA, Taylor JF, Van Tassell CP, Barendse W, Eversole KA, et al. Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science 2009;324(5926):528-532. [ Links ]
Recibido: 05 de Abril de 2018; Aprobado: 31 de Mayo de 2018
 This is an open-access article distributed under the terms of the
Creative Commons Attribution License
This is an open-access article distributed under the terms of the
Creative Commons Attribution License
