











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
Los miRNAs son secuencias de RNA de cadena sencilla, de 18 a 25 nucleótidos de longitud; en plantas, su biogénesis inicia cuando los genes MIRNAS son transcritos por la RNA polimerasa II, generando transcritos primarios con longitudes de 200 a 300 nucleótidos denominados como pri-miRNAs, que son procesados a una estructura única de tallo-burbuja por la acción de una ribonucleasa III (DCL), generando un dúplex de pre-miRNA; este dúplex es transportado del núcleo al citoplasma en donde se une con el complejo Argonauta (AGO) conformando el complejo RISC; finalmente, el complejo se une por complementariedad a los RNAs mensajeros objetivo, induciendo degradación o supresión de la traducción, por que los miRNAs son considerados moléculas silenciadoras de la expresión genética(1).
En plantas, estas moléculas participan en la regulación de diferentes etapas fisiológicas, como el cambio de la fase vegetativa a la floral, en el desarrollo y crecimiento de hojas, tallos, raíces y de órganos reproductivos. También se ha encontrado que los miRNAs regulan la respuesta de las plantas a distintas condiciones de estrés abiótico y biótico como la carencia de nutrientes, salinidad, sequia, estrés oxidativo, frio, radiación UV y por la presencia de virus2,3. Estudios in silico dirigidos a la ortología de los miRNAs en plantas, han demostrado que los miRNAs son conservados entre especies; sin embargo, también existen miRNAs específicos de especie4. Los miRNAs maduros en plantas son conservados en mayor grado que los pre-miRNAs2,5. La mayoría de los miRNAs que se encuentran conservados en distintas especies regulan genes homólogos5, esta característica permite analizar y predecir los posibles RNAs mensajeros blancos de miRNAs de especies que no tienen su genoma secuenciado. La predicción de mRNA blanco es indispensable para identificar la función del miRNA, por lo general, esto se realiza bioinformáticamente. El principal criterio usado es la detección de secuencias complementarias entre el miRNA y su blanco6; esto es posible ya que la complementariedad entre ellos es perfecta o casi perfecta, lo que permite una identificación de mRNAs blanco rápida y confiable2,7.
Bouteloua gracilis (Willd. ex Kunth) Lag. ex Griffiths es un pasto perenne perteneciente a la familia Poaceae con metabolismo C4 , de tallo delgado que crece en bosques, pastizales y matorrales 8,9. Se localiza en las regiones semiáridas de los Estados Unidos y México10. Esta gramínea es de gran importancia económica, ya que es fuente de forraje para la alimentación de ganado bovino y fauna nativa, por su alto contenido de proteína y alto grado de digestibilidad, además de su capacidad de adaptación a climas adversos como sequía y frío11,12. Debido a las características anteriores, este pasto ha sido sujeto de diversas investigaciones ecológicas y fisiológicas. Sin embargo, a la fecha no existe información acerca de los miRNAs expresados en B. gracilis y por lo tanto, se desconoce su relación con la regulación de la expresión genética en este pasto. En el presente trabajo se aislaron y caracterizaron secuencias de RNAs pequeños a partir de células clorofílicas de Bouteloua gracilis, con el objetivo de identificar miRNAs. Además, se realizó un análisis in silico para la identificación de los potenciales mRNAs blanco de los miRNAs encontrados, así como la búsqueda de los procesos biológicos en los que participan estos genes.
Material y métodos
Crecimiento de células clorofílicas de B. gracilis
La línea celular se desarrolló en INIFAP-Celaya13. Se agregó un inóculo de células clorofílicas de Bouteloua gracilis (H.B.K.) Lag. ex Steud., TIANSJ98, a 25 ml de medio MPC líquido, el cual contiene medio basal para plantas Murashige-Skoog, 2,4 -D, bencilaminopurina, adenina y sacarosa a pH de 5.8. Los cultivos se mantuvieron en agitación a 90 rpm, con luz continua fluorescente (77 μmols-1 m-2) a una temperatura de 33 ± 1 °C durante cinco días. Posteriormente, las células se cosecharon por filtración y se eliminó el exceso de medio. Se congelaron en nitrógeno líquido y se almacenaron a -70 °C hasta su uso.
Extracción de RNA total
Aproximadamente 150 mg de células de B. gracilis se lisaron por maceración con el sistema “Sample Grinding Kit™ (GE Healthcare, Suecia) y posteriormente, el RNA se aisló con el uso del método de extracción con Trizol™, (Ambion, EUA) y Plant RNA Isolation Kit™ (Ambion, EUA) de acuerdo a las especificaciones de los fabricantes.
Clonación de sRNAs
La clonación de los sRNAs por concatemerización se realizó con el kit miRCat™ (Integrated DNA Technologies, EUA) de acuerdo a las recomendaciones del fabricante. Los concatámeros obtenidos se clonaron en el vector PCR-TOPO 4 (Invitrogen™, EUA) para posteriormente introducirlo en bacterias químicamente competentes de E.coli One Shot TOP10™ (Invitrogen, EUA). A partir de las células transformadas, se realizó extracción de DNA plasmídico por minipreparación por lisis alcalina. Las secuencias de sRNAs insertadas en el sitio del polilinker del vector PCR-TOPO 4 se amplificaron con los oligonucleótidos T7™ (5’-TAA-TAC-GAC-TCA-CTA-TAG-GG-3’) y T3™ (5’-ATT-AAC- CCT-CAC-TAA-AGG-GA- 3’) que flanquean los sitios de clonación del vector. El programa de amplificación utilizado fue de desnaturalización inicial 94 °C por 1 min, 35 ciclos de desnaturalización a 94 °C por 1 min, alineamiento 55 °C 1 min, extensión 72 °C por 1 min y al finalizar los ciclos, una extensión final a 72 °C durante 10 min. Los productos de PCR se separaron en gel de agarosa al 1.5% y las bandas de 190 a 400 pares de bases (pb) se recuperaron del gel y se purificaron con el uso de minicolumnas Wizard SV Gel and Clean-Up System™ de acuerdo a las especificaciones del proveedor. El DNA amplificado y purificado se envió a la unidad de síntesis y secuenciación de DNA del Instituto de Biotecnología de la UNAM, ubicado en Cuernavaca, Morelos, México. La secuenciación se realizó en un equipo Perkin Elmer modelo 3730 (Applied Biosystems, EUA).
Análisis bioinformático
Las secuencias obtenidas se descargaron en formato FASTA y mediante análisis de alineamiento utilizando el programa BLASTn, a través de la plataforma del NCBI (http://blast.ncbi.nlm.nih.gov/Blast.cgi), con el algoritmo de alineamiento de dos o más secuencias, y parámetros preestablecidos para un alineamiento de alta similitud (Megablast). Se eliminaron secuencias correspondientes al vector, así como las de los conectores (5’-ACC-TTG-GTG - CCT- ACA-G -3’) presentes entre secuencias de los posibles miRNAs.
Las secuencias candidato de 18 a 25 nucleótidos se analizaron en la base de datos miRBase14, comparándose con los miRNAs maduros del reino Viridiplantae ahí reportados, de acuerdo al algoritmo y parámetros preestablecidos. Las secuencias candidatas también se analizaron en la plataforma UEA Workbench15 con la herramienta miRCat, para obtener la secuencia precursora, así como su estructura secundaria. Se usaron como referencia los genomas de Oryza sativa, Zea mays, Sorghum bicolor y Glycine max con los parámetros preestablecidos. Además, todas las secuencias se compararon con la base de datos Rfam16 para buscar posibles anotaciones dentro de los diferentes tipos de RNAs (rRNAs, tRNAs, mRNAs, ncRNAs, etc.) con los parámetros preestablecidos.
Finalmente, a las secuencias que tuvieron similitud con miRNAs conocidos se les realizó la búsqueda de mRNA objetivo con el programa psRNATarget17. Debido a que el genoma de Bouteloua gracilis no se ha reportado, se utilizó la base de datos de anotación de transcritos de arroz MSU versión 7 18. Los parámetros usados para la búsqueda mRNAs objetivo fueron más restrictivos que los preestablecidos, el “E value” (máxima expectación) máximo usado fue 2.0 (3.0 preestablecido) y el valor “UPE” (accesibilidad a la región de unión) como máximo 20 (25 preestablecido); al disminuir los valores máximos de ambos parámetros se obtienen menos falsos positivos17. Se descargaron las secuencias de los genes objetivo potenciales, y se realizó un análisis funcional de estos, con el programa Blast2GO19.
Resultados
Identificación de miRNAs en B. gracilis
De 10 plásmidos que fueron amplificados, se seleccionaron cuatro que generaron productos de PCR en un rango de 400 a 150 nucleótidos. La secuenciación y análisis del DNA amplificado demostró la existencia de nueve secuencias con longitudes de 18 a 25 nucleótidos característicos de los miRNAs (Cuadro 1). Estas secuencias se analizaron en la base de datos Rfam, observándose la ausencia de similitud con los RNAs ahí reportados (rRNA, tRNA snRNA, snoRNA). El análisis de homología de las secuencias con los miRNAs depositados en miRBase14, demostró que las secuencias uno a cuatro tuvieron el mejor alineamiento con los miRNAs miR7828-5p de Populus trichocarpa20; el miR319 reportado en Brachypodium distachyon21, Oryza sativa22, Sorghum bicolor23 y en Zea mays24; el miR397-3p de Malus domestica25 y el miR397-5p reportado en Linum usitatissimum26 y en Brachypodium distachyon23 (Cuadro 1). Sin embargo, las secuencias cinco a nueve, no mostraron similitud con miRNAs reportados previamente.
Cuadro 1 Análisis in silico de las secuencias de sRNAs obtenidas a partir de concatámeros de B. gracilis
| No | Sequence 5'-3' | Lg | miRBase | Species | ||
| 1 | GATCAGATGGAGGCTAAAATC | 21 | miR7828-5p | Ptr | ||
| 2 | AGGGAGCACCCTTCAGTCCAA | 21 | miR319 | Bdi Osa Sbi Zma | ||
| 3 | TTTCATCAACGCTGCACTCAA | 21 | miR397-3p | Mdm | ||
| 4 | ATTGAGTGCAGCGTTGATGAA | 21 | miR397-5p | Lus Bdi | ||
| 5 | ACCAACTAACCTTAGGCA | 18 | NF | NA | ||
| 6 | AAACCCCAGTTTCACACCTCCGCT | 24 | NF | NA | ||
| 7 | GTACACACATCGATCAGTCTTATT | 24 | NF | NA | ||
| 8 | TGTACATCACTAAGGTAACAAAA | 23 | NF | NA | ||
| 9 | CAATGACATCGCAAACAAGTGCCT | 24 | NF | NA | ||
MiRBase database14 was used to identify mature miRNAs.
Lg= Length of the sequence; Ptr= Populus trichocarpa; Bdi= Brachypodium distachyon; Osa= Oryza sativa; Sbi= Sorghum bicolor; Zma= Zea mays; Mdm= Malus domestica; Lus= Linum usitatissimum.
NF=Not found; NA= Not apply.
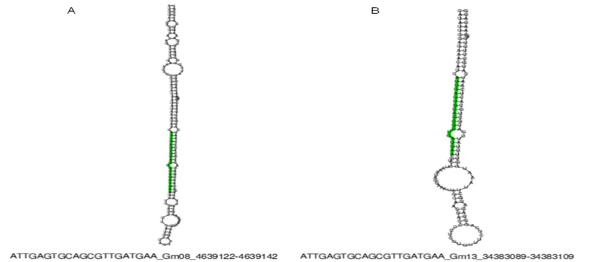
Con el fin de caracterizar estas secuencias, se utilizó la herramienta miRCat de la plataforma UEA Workbench 15 que tiene como función la predicción de miRNAs nuevos, ya que realiza un mapeo de los potenciales miRNAs en estudio a un genoma de interés, y en caso de que el mapeo sea positivo a dicho genoma, determina si el miRNA tiene una secuencia precursora con estructura secundaria tallo- burbuja (pre-miRNAs). El análisis se realizó con todas las secuencias obtenidas y los resultados demostraron que sólo los miRNAs ya caracterizados (miR397 y miR319) generaron pre-miRNAs. Así la secuencia dos (miR319) se localizó en los genomas de Zea mays, Oryza sativa y Sorghum bicolor donde se obtuvo la estructura de su pre-miRNA (Cuadro 2; Figura 1). Respecto a la secuencia cuatro (miR397-5p) también fue posible identificar su estructura secundaria, pero únicamente en el genoma de Glycine max (Cuadro 2; Figura 2). Respecto a las secuencias uno, cinco, seis, siete, ocho y nueve, no fue posible identificarlas en ninguno de los genomas de referencia.
Cuadro 2 Análisis in silico de las secuencias de sRNAs obtenidas a partir de concatámeros de B. gracilis, con la plataforma miRCat15, con los genomas de soya, maíz, sorgo y arroz
| Spe | Loc | Start | End | St | Sec 5'-3' | Lg | Lg Pre | % G/C | MFE |
| Zma | chr3 | 210558024 | 210558044 | - | AGGGAGCACCCTTCAGTCCAA | 21 | 170 | 54 | -66.5 |
| Osa | Os01g46984 | 541 | 561 | - | AGGGAGCACCCTTCAGTCCAA | 21 | 192 | 50 | -80.1 |
| Sbi | chro_3 | 1240353 | 1240373 | - | AGGGAGCACCCTTCAGTCCAA | 21 | 200 | 56 | -94.3 |
| Gma | Gm08 | 4639122 | 4639142 | - | ATTGAGTGCAGCGTTGATGAA | 21 | 179 | 39 | -67.3 |
| Gma | Gm13 | 34383089 | 34383109 | - | ATTGAGTGCAGCGTTGATGAA | 21 | 136 | 37 | -56.4 |
Spe= Species; Loc= location in the genome; Start= Location of start of sequence in the genome; End= Location of end of the sequence in the genome; St= string where sequence is located: Sec 5'- 3‘= sequence in the 5’ to 3 ‘ direction; Lg= length of the sequence; Lg Pre= length precursor sequence; %G/C= percentage of the content of guanine and cytokines in the pre-miRNA sequence; MFE= minimum free energy of the structure of pre-miRNA; Zma= Zea mays; Osa= Oryza sativa; Sbi= Sorghum bicolor; Gma= Glycine max; Gm08= Cromosome 8; Gm13= Cromosome 13; chro_3= Cromosome 3; Os01g46984= location number of the rice genome sequence.
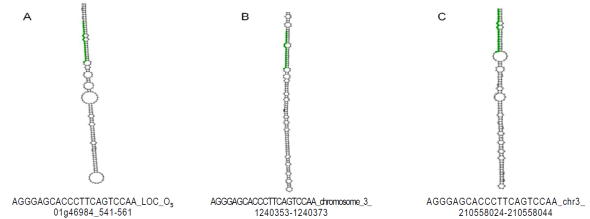
Figura 1 Estructura secundaria de los pre-miRNAs predichos bioinformáticamente para la secuencia dos (miR319-3p). En verde, se señala la secuencia del miRNA identificado. A) Localización en el cromosoma uno de Oryza sativa. B) Localización en el cromosoma tres de Sorghum bicolor. C). Localización en el cromosoma tres de Zea mays
Identificación de RNAm blancos
Con el uso del programa psRNATarget17 que predice mRNAs objetivo con base en el grado de complementariedad y en la capacidad de acceso a la región complementaria, se procedió a la identificación de los posibles blancos de todas las secuencias. El análisis demostró que sólo a miR397-5p fue posible predecirle seis mRNAs objetivo, los cuales codifican para proteínas lacasas (Cuadro 3). El análisis funcional de los genes blanco demostró que las proteínas que codifican participan en procesos metabólicos de lignina y procesos catabólicos de fenilpropanoides. Además de estas funciones, cada gen tiene anotación distinta involucrada en otros procesos. Cinco (Os01g63190, Os01g63200, Os05g38420, Os11g48060 y Os12g15680) tienen anotación de unión a Cobre. Dos (Os01g63200 y Os12g15680) participan en el desarrollo del sistema reproductivo. Mientras que Os11g48060 tiene funciones involucradas en la formación de pared celular y Os05g38420 participa durante la respuesta por falta de agua.
Cuadro 3 mRNAs blanco predichos para la secuencia cuatro (miR397-5p) identificados con el programa psRNATarget17
| Name Target | Ev | UPE | Target start | Target end | Target Alignment 5'-3' | Target Description |
| Os05g38420 | 1.0 | 17.7 | 746 | 765 | UCAUCAACGCUGCACUCAAC | Protein Precursor laccase |
| Os12g15680 | 1.5 | 13.1 | 748 | 767 | UCAUCAACGCUGCGCUCAAC | Protein Precursor laccase |
| Os01g63180 | 1.5 | 17.4 | 1087 | 1106 | UCAUCAACGCUGCGCUCAAC | Precursor laccase6 |
| Os01g63200 | 1.5 | 19.7 | 665 | 684 | UCAUCAACGCUGCAGUCAAU | Protein Precursor laccase |
| Os11g48060 | 1.5 | 14.5 | 1179 | 1198 | UCAUCAACGCUGCACUGAAU | Precursor laccase 22 |
| Os01g63190 | 2.0 | 17.5 | 753 | 772 | UCAUCAACGCUGGACUCAAC | Protein Precursor laccase |
Database annotation of rice transcripts MSU version 718) was used as reference.
Ev= Expectation value; UPE= Accessibility to the target value.
Discusión
La obtención de secuencias similares a miRNAs maduros reportados en la miRBase indica que Bouteloua gracilis sintetiza estas moléculas para la regulación de su expresión genética. De las nueve secuencias aisladas, sólo cuatro tuvieron similitud con miRNAs conservados en la miRBase; mientras que en las cinco restantes, no se encontraron registros de secuencias ortólogas en otras especies de plantas. El análisis de mapeo con el programa miRCat de nuestros potenciales miRNAs, con genomas de otras especies de plantas demostró clara correlación de los miRNAs reportados en la miRBase y su mapeo en los genomas de las especies en las que fueron identificadas, así como la generación de estructuras secundarias tallo-burbuja estables; sin embargo, el resto no mostró mapeo a ningún genoma seleccionado. En conjunto, estos resultados confirman el alto grado de conservación de los miRNAs, miR319 y miR397 encontrados en gramíneas como Zea mays, Oryza sativa y Sorghum bicolor, además de la leguminosa Glycine max, así como en B.gracilis. Es interesante comentar que aunque la cantidad de secuencias obtenidas es baja, fue posible detectar la presencia tanto del miR397-5p, como de su complementario miR397-3p, por lo que es posible que este miRNA tenga participación activa en B. gracilis. Con respecto a las secuencias cinco a nueve, la ausencia de ortólogos así como el mapeo negativo a alguno de los genomas de referencia empleados, indica la posibilidad de que estas secuencias sean específicas a B.gracilis. Respecto a la secuencia uno, a pesar de que tuvo similitud con el miR7828-5p de Populus trichocarpa, ésta fue muy baja, ya que tuvo cinco incomplementariedades.
Los potenciales mRNAs blanco detectados fueron predichos a partir de la secuencia cuatro (miR397 -5p); por tanto, es posible que la expresión de este miRNA regule de manera negativa la expresión de las lacasas, que son enzimas glicoprotéicas que tienen la función de oxidar compuestos fenólicos presentes en la lignina27. La inhibición de la expresión de estas enzimas puede ser debida al hecho de que, como las células clorofílicas son indiferenciadas, en las que no existe la presencia de lignina, no se requiere la presencia de lacasas para que oxiden a dichos compuestos fenólicos. O bien, al no haber lignina presente, las lacasas pudieran oxidar otro tipo de compuestos fenólicos, lo que podría afectar el buen funcionamiento celular. Además, estos genes también tuvieron anotaciones funcionales involucradas en el desarrollo del sistema reproductivo, en formación de pared celular y respuesta a falta de agua, por lo que también es de esperarse que estos genes estén siendo silenciados, ya que es posible que ninguno de estos procesos biológicos ocurra en las células indiferenciadas, a partir de las cuales se realizó este estudio. Por otro lado, el miR319 es un miRNA muy caracterizado, y aunque no fue posible predecirle algún blanco en este análisis, se ha reportado que tiene como objetivos principales los factores de transcripción TCP clase II; los cuales, participan en la regulación negativa del crecimiento celular reportado en Arabidopsis28.
El hecho de que el resto de secuencias no presentaran posibles blancos no significa que estos no existan; lo anterior, se puede deber a que los parámetros seleccionados para realizar el análisis fueron muy elevados para evitar al máximo falsos positivos, o que los blancos sean secuencias específicas de B. gracilis.
Conclusiones e implicaciones
En el presente estudio fue posible identificar cuatro miRNAs reportados en miRBase; dos de ellos, altamente conservados. Estos resultados confirman la presencia de RNAs pequeños como los miRNAs en B. gracilis . La identificación del miR397-5p, miR397-3p, así como sus potenciales mRNAs objetivo, que codifican a enzimas con actividad de lacasas, es un indicio de que esta molécula participa muy activamente en estas células. La evaluación de la expresión de este miRNA en diferentes tejidos de la planta, así como en células y plantas desarrolladas bajo condiciones de estrés hídrico, permitirá conocer la relevancia fisiológica que tiene este miRNA en B gracilis.

