











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
Una de las grandes virtudes de la sabiduría del riñón es su gran capacidad adaptativa a la destrucción progresiva de sus unidades funcionales o nefronas.
Hace muchos años el maestro Ignacio Chávez, clínico sagaz y médico irrepetible, en una de sus pláticas memorables comentó con su rica oratoria: “El paciente cardiaco va siempre mal, mal, mal, hasta que se muere; por el contrario, el paciente renal va bien, bien, bien, hasta que se muere”.
¿Qué secretos posee el riñón para que con pérdidas hasta de más de 80% de su capacidad funcional, el enfermo se mantenga activo y casi asintomático hasta que violentamente desarrolla un cuadro florido de uremia? Pero si no se toman medidas de tratamiento drásticas, lleva al paciente a una muerte segura, violenta, convulsa y aterradora. Cuando era residente en el viejo Hospital de Enfermedades de la Nutrición, en ausencia de métodos de tratamiento adecuados, me enfrenté a pacientes con esta terrible enfermedad que morían ante nuestros ojos sin poder hacer casi nada por ellos.
En esa época, ya aficionado a las enfermedades del riñón, pero más que nada a su fisiología, leí con avidez el gran libro del Dr. Homer W Smith The Kidney in Health and Disease,1 donde me inicié en el estudio de este órgano fascinante (1955). Con estos antecedentes decidí especializarme en esta naciente área de la medicina, la nefrología, pero antes que nada estudiar fisiología renal. Mi primer amor fue el sodio y el notable control del “medio interno”. Mi primer modelo de estudio fue el paciente cirrótico con ascitis que estudié con mi amigo, mi jefe y mi maestro, el Dr. Alfonso Rivera Valenzuela en la primitiva Unidad Metabólica del Hospital de Enfermedades de la Nutrición enclavado en la Colonia de los Doctores. Rivera, alumno de Frederic C Bartter,2 el gran gurú del balance metabólico y de los trastornos relacionados con la sal y el agua, sin duda uno de los más grandes investigadores clínicos de su tiempo, indirectamente fue mi maestro a través de Rivera. Esos tiempos fueron ricos en la adquisición de conocimientos que me permitieron adentrarme en la capacidad del riñón para controlar el “medio interno”.
Cuando llegué a los EUA, primero al Departamento de Fisiología de la Universidad de Michigan en Ann Arbor, el bellísimo pueblo universitario, con su jefe el Dr. Horace W Davenport, a la sazón presidente de la Sociedad Americana de Fisiología, con mi jefe directo y maestro el Dr. Richard L Malvin, PhD en fisiología. Era un departamento en expansión con grandes investigadores en todos los campos de la fisiología. Esta estancia me permitió aumentar aún más mis conocimientos sobre mi órgano amado: el riñón, pero también en otras áreas y metodologías.
De ese lugar de excelencia pasé a otro de superexcelencia, el Renal Laboratory en el Peter Bent Brigham Hospital y la Universidad de Harvard, con su jefe el Dr. John P Merrill, pionero de los trasplantes y la diálisis. Hombre carismático, acababa de salir en la portada del Times Magazine por sus primeros trasplantes de riñón en gemelos idénticos y no idénticos. A ese lugar de excelencia llegué en el año de 1960.
En uno de nuestros seminarios de ese tiempo me tocó revisar un artículo escrito por el gran patólogo renal, el Dr. Jean Oliver, su título “When is the kidney not a kidney”.3 En este artículo el Dr. Oliver asentaba, lo traduzco literalmente. “La respuesta que yo daría a esta pregunta, actualmente bastante obvia, es que no hay “riñón” ni estructural ni funcionalmente en la enfermedad renal crónica”. Con su retórica decimonónica el Dr. Oliver nos decía que el riñón con daño renal crónico, aun cuando pequeño y con cicatrices, mantenía su aspecto de riñón. En estas condiciones, cuáles serían las capacidades funcionales de ese riñón crónicamente enfermo. Sería lógico concluir que la profunda alteración histológica del riñón debería asociarse a una gran desorganización funcional, y de hecho, ésa fue la conclusión de muchos morfólogos de la época encabezados por el Dr. Oliver. La extrapolación de estructura a función les pareció lógica a los fisiólogos y parecía tener fundamento en el hecho de que la función renal en la uremia difería de manera importante de la función renal normal.
A pesar de la renuencia del Dr. Oliver de aceptar los métodos de depuración ampliamente difundidos por la escuela del Dr. Homer W Smith1,4 para estudiar el riñón enfermo, éstos se aplicaron con éxito en su estudio y su relación órgano-función.
Es interesante mencionar que el Dr. Oliver al final de su ensayo afirma que las alteraciones en el riñón afectan directamente a las nefronas y que de hecho su próximo ensayo se podría titular “Las nefronas en la salud y la enfermedad”. En los años siguientes a este trabajo un sinnúmero de investigadores, pero especialmente el Dr. Neal Bricker con su grupo en San Louis Missouri, se lanzó al estudio de la enfermedad renal crónica con métodos de depuración. En uno de sus artículos publicado en 1960 planteaba la hipótesis de la nefrona intacta.5 Los resultados que obtuvo fueron de gran trascendencia en la comprensión del daño renal y de la gran capacidad de adaptación del riñón que permite una sobrevida libre de síntomas como relataremos a continuación. Ya unos años antes el Dr. Platt en Inglaterra había sugerido este comportamiento del riñón.6
Esta facultad de los animales superiores de mantener su medio interno constante es responsabilidad de los riñones, aun en la insuficiencia renal.
“Se puede afirmar sin exageración que la composición y el volumen de los líquidos corporales no es el resultado de los que ingerimos, sino de lo que los riñones son capaces de retener. Son ellos, ese conglomerado de vasos, túbulos, vénulas y linfáticos, los verdaderos reguladores del medio interno. Su capacidad excretora, que se caracteriza por eliminar todo aquello que en forma por demás indiscriminada absorbemos del tubo digestivo o desechamos de las células hacia el torrente circulatorio, constituye su labor primordial: mantener en un estado de balance ideal nuestro medio interno”. Homer W Smith en su libro From Fish to Philosopher.7
La magnitud de adaptación de los riñones es tal, que la composición de los líquidos corporales no se modifica en la enfermedad renal hasta que se ha perdido más de 85 % de su función.
La hipótesis de la nefrona intacta
A partir de 1960 el grupo del Dr. Bricker5,8,9 llevó a cabo numerosos estudios que lo condujeron a la conclusión de que: la nefronas que sobreviven al embate de la enfermedad renal crónica, retienen su integridad funcional sin importar el origen del daño renal. Hay situaciones en que las alteraciones histológicas son compatibles con algunas alteraciones funcionales, como se verá más adelante, pero éstas son poco frecuentes.
La reducción de 5/6 de la masa renal mediante nefrectomía en ratas o perros sanos deja intactas y sanas las nefronas remanentes. Éste ha sido y es un modelo experimental muy utilizado. Las nefronas remanentes de los animales con 5/6 de nefrectomía son funcionalmente sanas, sólo sufren dilatación e hipertrofia. Estos hallazgos sugirieron que las alteraciones funcionales en este modelo son semejantes a la de un riñón enfermo (nefritis de Heyman y otros modelos). Lo que sugería que las nefronas remanentes en el riñón enfermo también permanecían intactas. De hecho, en experimentos con micropunción en túbulos proximales en modelos animales con daño renal, la relación de inulina del fluido tubular proximal con respecto al plasma (TF/P inulina) era igual a la de los animales sanos o con 5/6 de nefrectomía.5,8
¿Cómo funciona el riñón enfermo?
Para estudiar las funciones del riñón enfermo se empleó un modelo con lesión renal unilateral en el perro y en el humano sin uremia.8
Los resultados de la medición del riñón enfermo en el perro en comparación con el riñón sano se muestran en la Figura 1. Se estudiaron ambos riñones, el control y el experimental, antes y después de la lesión. La filtración glomerular (FG) se estudió con depuración de inulina. En la parte superior de la gráfica se observa la función renal de los dos riñones normales, uno el que se va a lesionar, en oscuro. En la parte derecha, se observa el descenso de la FG en el riñón enfermo, en oscuro, al compararlo con el riñón sano, en claro. En la parte de abajo se muestra este mismo diseño, pero se grafican diversas funciones tubulares corregidas por la FG. La primera, la fracción filtrada, al dividir flujo plasmático renal efectivo entre la FG (FPR/FG), que se mantiene igual tanto en el riñón sano como en el enfermo. Además, se presentan otras funciones tubulares como transporte máximo de paramino hipurato de sodio (Tm PAH), Tm de glucosa y fosfato así como la excreción urinaria (OV) de urato, amonio y acidez titulable que, al corregirse por la FG, son iguales tanto en el riñón sano como en el enfermo (Figura 1). Estos resultados apoyaban la homogeneidad de las funciones tubulares y excretoras de las nefronas remanentes y la gran capacidad adaptativa del riñón a la progresión del daño renal; asimismo indicaban claramente que las nefronas que determinaban la función estaban intactas, y que las nefronas parcial o totalmente destruidas no contribuían a la función renal total; y si lo hacían, sería en un porcentaje tan bajo que no modificaban los resultados. Los datos de micropunción en modelos de 5/6 de nefrectomía o de daño renal en ratas y perros contribuyeron aún más a reafirmar la hipótesis de la nefrona intacta.8
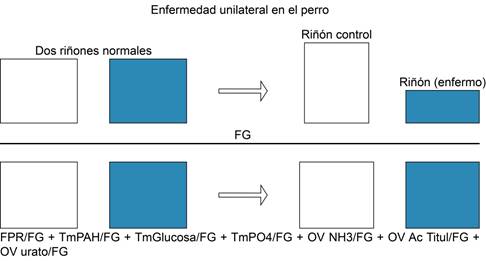
Figura 1: Relación funcional entre dos riñones en el perro. El cambio en la filtración glomerular en ambos riñones después de la inducción de enfermedad renal se muestra en las columnas de la parte superior de la gráfica. En las columnas inferiores se muestran los valores de varias funciones tubulares corregidas por la filtración glomerular que son iguales en el riñón sano y en el enfermo. Tm representa el transporte máximo de paramino hipurato de sodio (Tm PAH), glucosa y fosfatos, excreción urinaria es la excreción/min de NH3, Ac titulable y uratos. FPR es el flujo plasmático renal efectivo. FPR = flujo plasmático renal; TM PAH = transporte máximo de paramino hipurato de sodio; Tm Glucosa = transporte máximo de glucosa; Tm PO4 = transporte máximo de fósforo; OV de amonio = excreción de amonio; FG = filtración glomerular; Ac titul = acidez titulable. Tomado y modificado: Bricker NS et al.8
Excreción de sodio
El paciente renal crónico con grandes déficits de función renal mantiene un balance de sodio casi perfecto hasta estadios muy avanzados de la enfermedad renal.10 En la Figura 2 y Tabla 1 se ejemplifica este comportamiento en el que se observa que en la medida en que la función renal decrece, en ese mismo porcentaje aumenta la excreción de sodio. Por ejemplo (Tabla 1), con 2,000,000 de nefronas, filtramos en 24 horas 17,300 mEq de Na, excretamos en la orina 173 meq/día, por lo que la fracción excretada de sodio (FeNa) es 1%. Si se reduce el número de nefronas a la mitad (1,000,000), la fracción filtrada de Na disminuye 50 %, es decir, a 8,650 mEq/día y para excretar la misma cantidad de Na que en el sujeto normal (173 Meq), la FeNa debe aumentar a 2%. Si el número de nefronas se reduce aún más a 250,000, la FeNa aumenta a 8% y el paciente se mantiene en balance de sodio (Tabla 1). Esta capacidad del riñón para mantener el balance de sal y agua permite al enfermo evolucionar sin edema aparente y asintomático con pérdidas de más de 80% de la función renal (Figura 2). Es muy importante mencionar en este punto que si se reducía la ingestión de sodio a prácticamente cero, la excreción de sodio se mantenía en balance negativo, lo que sugirió a los investigadores en esa época (1960) que había un defecto tubular para reabsorber sal. Años después de aceptar la presencia de este defecto, el Dr. Bricker hizo una visita a mi departamento y en un seminario nos comentó: “Acabo de hacer una observación por demás interesante. En pacientes con riñón poliquístico con daño renal crónico avanzado sometidos a una dieta muy restringida en sodio, al cabo de 12-15 días todos ellos entraron en balance de sal sin reducir sustancialmente su función renal residual. Esto indicaba que, por lo menos en estos enfermos, no existía un trastorno tubular con pérdida de sodio”. En los siguientes años se dilucidó que este comportamiento de la excreción de sodio ocurría en todos los pacientes con daño renal crónico, no importaba la etiología; el único requisito para llegar al balance con una dieta prácticamente sin sal era el tiempo. El sujeto normal en cuatro días de dieta baja en sodio entraba en balance, mientras que el paciente renal tardaba más días.
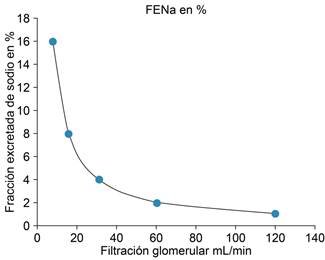
Figura 2: Manejo hipotético de la excreción de sodio en la enfermedad renal crónica. FENa % = excreción fraccional de sodio en %.
| No. de nefronas | FG mL/min | FF Na meq/día | FENa en % |
|---|---|---|---|
| 2,000,000 | 120.0 | 17,300 | 1 |
| 1,000,000 | 60.0 | 8,650 | 2 |
| 500,000 | 30.0 | 4,325 | 4 |
| 250,000 | 15.0 | 2,118 | 8 |
| 125,000 | 7.5 | 1,059 | 16 |
En esa misma época en Inglaterra, el Dr. Hugh De Wardener y su grupo11 realizaban una serie de experimentos muy trascendentales que se caracterizaban por retar a animales de laboratorio tratados con arginina vasopresina y aldosterona con cargas salinas agudas que producían un gran aumento en la fracción excretada de sodio. Esto sugería que en la expansión del espacio extracelular, otros factores hormonales o ambos eran los responsables de esta gran natriuresis. A este estudio se sumó el llamado escape a la DOCA. Sujetos normales tratados con este mineralocorticoide en dieta normal en sal; los primeros días retenían sodio y entre el quinto y el séptimo día escapaban a su efecto y la excreción de sodio igualaba a su ingestión.12
Como se observa en la Figura 2 y Tabla 1, la excreción de sodio aumenta en proporción a la reducción de nefronas y a la caída de la FG. La explicación a este fenómeno es que a medida que se reduce la función renal, en esa misma proporción el enfermo renal retiene sal, el volumen extracelular se expande y la fracción excretada de sodio aumenta. Tal como ocurre en el sujeto normal al expandir su volumen corporal, particularmente el espacio extracelular, con soluciones salinas o retarlo con dietas altas en sodio o con DOCA.11,12 Es una oferta de trueque (trade off), que es el mecanismo mediante el cual el organismo enfermo mantiene el balance de sodio a expensas de una retención progresiva de sodio y agua. Esta expansión del espacio extracelular se acompaña de la inhibición del sistema renina-angiotensina-aldosterona y de aumento del factor natriurético auricular, mecanismos ambos que mantienen en balance la excreción de sal. Más adelante discutiremos más ampliamente esta hipótesis del trueque o trade off, cuando analicemos la fisiopatología del hiperparatiroidismo secundario en la insuficiencia renal.
Elevación de urea y creatinina
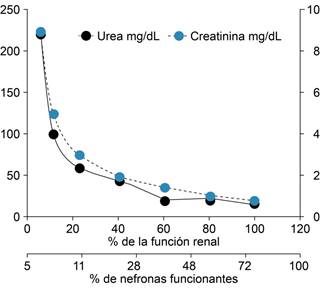
Otro ejemplo de esta adaptación es cómo se incrementan solutos como la urea y la creatinina a medida que se pierde la función renal.13 Como se ejemplifica en la Figura 3, se trata de un incremento exponencial de ambas variables urea y creatinina. Lo interesante de estas curvas es que son tan parecidas que las grafiqué juntas. Ambos metabolitos se mantienen en valores normales o muy cercanos a ellos hasta que la pérdida de la función renal va más allá de 80%. A este descenso en la función se suman otras alteraciones bioquímicas, minerales, endocrinas, hematológicas, nutricionales y tóxicas. Éstas son progresivas y todas sumadas constituyen clínicamente lo que conocemos como síndrome urémico.
Concentración urinaria
Otra característica del enfermo renal es que la concentración urinaria es limitada desde las etapas iniciales del daño renal. El ciclo circadiano de la diuresis está alterado con nicturia y poliuria tardía de no más de 3-4 litros. La orina del enfermo renal es isostenúrica. Hay tolerancia a las cargas de agua, pero si éstas son en exceso, el paciente puede desarrollar hiponatremia. Lo que se acepta en la actualidad es que la inducción de diuresis de agua y el bloqueo completo de la hormona antidiurética (HAD) protegen contra la progresión del daño renal. La fisiopatología de esta limitación en la capacidad para concentrar la orina depende de que las nefronas remanentes estén sometidas a una carga osmolar excesiva que reduce el gradiente osmótico del intersticio en la nefronas yuxtamedulares y reduce la concentración urinaria.14,15 A esto se suma una resistencia al efecto de la HAD. Esta limitación en la concentración urinaria ocurre también en sujetos sanos sometidos a diuresis osmótica.
Manejo del potasio
El potasio tiene como característica fisiológica que se filtra y se reabsorbe casi en su totalidad y su excreción es el resultado de la secreción del ion en las porciones distales de la nefrona, particularmente el tubo colector. En el riñón insuficiente este mecanismo se conserva y la secreción distal de K aumenta en las nefronas intactas. Un estudio realizado en Japón16 en 989 pacientes en diferentes estadios de daño renal de G1 a G5 se muestra claramente en las Figura 4, se observa que la excreción de K se reducía a medida que progresaba el daño renal. En los estadios de 1 a 3b no hay cambios significativos en la excreción, el K desciende discretamente en el estadio 4 y claramente en el estadio 5 si se compara la excreción entre los estadios G1 a G3b versus el G4 y contra G5. En la Figura 5 se observa la fracción excretada de K en porciento (%) en la medida que la función renal decrece en los diversos grupos, el porcentaje de excreción de K aumenta hasta ser máxima en el estadio 5. El K sérico se mantiene en límites normales a lo largo de la enfermedad renal aun en los estadios 4 y 5.16 Sin embargo, si el sujeto es sometido a una carga excesiva de K en la dieta o por administración endovenosa así como por el empleo de drogas que inhiben el sistema renina-angiotensina o directamente la aldosterona, pueden ocurrir incrementos en el K en sangre que pongan en peligro la vida.

Figura 4: Excreción de potasio en pacientes con daño renal progresivo en meq/día. * p < 0.05, ‡ p < 0.01. Modificada de: Ueda Y et al.16

Figura 5: Excreción fraccional de K en estadios progresivos de daño renal. * p < 0.01. Modificada de: Ueda Y et al.16
La hipótesis de la nefrona intacta en el contexto del manejo del calcio y fósforo y los trastornos del metabolismo mineral.
La alteración fundamental que ocurre especialmente en el metabolismo del fósforo, es que se retiene desde etapas tempranas del daño renal. Sin embargo, sus valores se mantienen en cifras normales de 3.5 a 4.0 mg/dL hasta que, con pérdidas de función renal de más de 80%, el fósforo en suero tiende a elevarse. Tal como se observa en la Figura 6.17
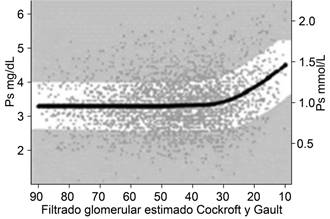
Figura 6: El fósforo sérico se mantiene en límites normales hasta pérdidas de casi 80% de la función renal. N = 3,490. Modificado de: Kestenbaum B et al.17
Los valores de calcio tampoco varían. Este comportamiento del fósforo dio lugar a la hipótesis de la nefrona intacta y del trueque (trade-off). En la Figura 7, diseñada por el Dr. Bricker en la década de los 70, se ejemplifica cómo a medida que el fósforo se incrementaba por caída de la función renal, la hormona paratiroidea (PTHi) se elevaba, este aumento producía fosfaturia que corregía automáticamente el fósforo de la sangre y a cambio de eso la PTH se mantenía elevada.

Figura 7: En este esquema se observa el aumento de la PTH que permite la corrección del fósforo hasta que la filtración glomerular cae a valores menores de 20 mL/min.
Este proceso se repetía a medida que se reducía la función renal y la PTH se mantenía cada vez más elevada, tal como se observa en la Figura 7. Por el contrario, el calcitriol o 1,25, dihidroxicalciferol (Vit D3) reducía sus valores en sangre a medida que se elevaba la PTHi (Figura 8). Con estos dos esquemas el Dr. Bricker le dio más fuerza a su “hipótesis del trueque” y de las nefronas intactas.8,9,18
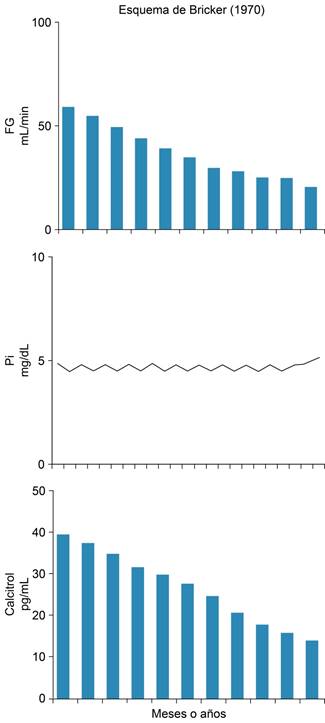
Figura 8: En este esquema se observa el descenso del calcitriol a medida que cae la función renal y se eleva la PTH.
En la actualidad se añadió al esquema de la PTHi la FGF 23, que es una hormona sintetizada en los osteocitos del hueso y que es intensamente fosfatúrica y añade su efecto al de la PTH en la corrección progresiva del fósforo, tal como se observa en la Figura 9.18
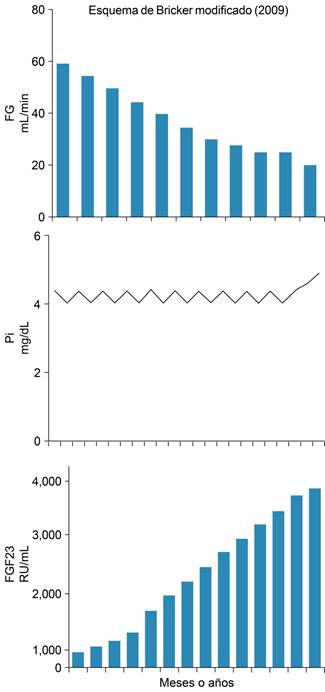
Figura 9: Se muestra el aumento en FGF23 que se eleva a medida que desciende la filtración glomerular, semejante a lo que ocurre con la PTHi.
En resumen, la corrección progresiva del fósforo en la magnitud que la función renal disminuye, depende de tres factores hormonales, la PTH y la FGF23, ambas con un gran efecto fosfatúrico, al que se suma el descenso progresivo del colecalciferol o vitamina D3 que reduce la absorción intestinal de fósforo (Figuras 7 a 9). Estos mecanismos mantienen el fósforo en límites normales, tal como se observa en la Figura 6, hasta que en etapas avanzadas de la lesión renal con pérdidas de más de 80% de la función renal, los mecanismos de compensación son insuficientes y el fósforo se eleva (Figuras 6 a 9). Una observación interesante es que la administración de quelantes del fósforo y dietas bajas en este elemento en perros con daño renal, al evitar el ascenso del fósforo sérico se corrige la elevación de la PTH y el FGF 23, y el calcitriol incrementa sus valores.18 Esta maniobra es importante porque el trueque propicia la aparición de hiperparatiroidismo secundario con sus secuelas óseas como osteodistrofia renal y la aceleración del daño cardiaco, y el empleo de quelantes y de calcitriol o de sus análogos (paracalcitiol, alfa calcidiol) previenen estas secuelas óseas tan graves. La Figura 10 ejemplifica un grupo de pacientes en hemodiálisis tratados con un análogo del calcitriol el alfacalcidiol (1-alfa-hidroxicolecalciferol) por siete meses, con su administración los valores de PTHi se redujeron de 3,000 a 300 pg/mL y el calcitriol se elevó de 3.8 pg/mL a cifras normales de 48 pg por mL. La PTHI descendió a valores que evitan la progresión del daño óseo y cardiovascular.19
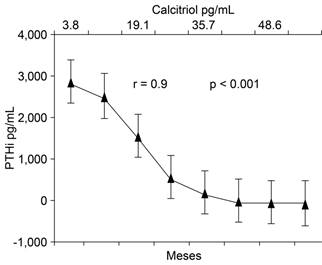
Figura 10: Correlación entre PTHi y calcitriol en suero en pacientes con IRC en diálisis tratados con alfacalcidiol. Modificado de: Peña JC et al.19
Equilibrio ácido-básico
El riñón posee tres mecanismos para excretar la carga ácida del organismo, la reabsorción de bicarbonato y la regeneración del bicarbonato consumido por la carga ácida normal o excesiva. Esta regeneración del bicarbonato consumido se hace a través de la excreción neta de ácido por dos mecanismos: la acidez titulable al excretar el H+ unido a sales de fosfato y de sulfato y la excreción de amonio. Se estima que de 25-50% de los 70 meq de H+ generado diariamente se excretan como acidez titulable y la misma cantidad en forma de amonio. Cuando se somete a un sujeto normal a una carga ácida excesiva, la excreción de amonio se eleva considerablemente (de cinco a 10 veces más que su excreción normal) y la acidez titulable sólo aumenta discretamente.20 En los años 70 el Dr. Bricker8 estudió la excreción de amonio en ambos riñones de perros, primero sanos (estadio I) y en ellos mismos después de inducir enfermedad renal unilateral (estadio II) y finalmente con nefrectomía del riñón sano (estadio III). En los tres estadios se mantuvo el mismo grado de acidosis metabólica, se les sometía a una carga de alanina precursor de la síntesis de amonio, que al metabolizarse lo incrementaba. Sus resultados fueron por demás interesantes, los riñones controles, uno de los cuales en el estadio I fue denominado experimental, se muestran en la Figura 11; se graficó la excreción de amonio después del reto con alanina. Esta excreción de amonio en μg se corregía en cada estadio por la FG, de esa manera la excreción de amonio permitía comparar los riñones sanos con los enfermos.
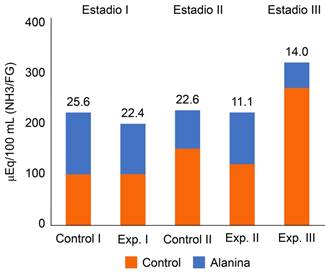
Figura 11: Excreción de amonio en perros, corregida por la FG, cuyos valores se representan en la parte superior de las barras; en los tres estadios del estudio. Tomada y modificada de: Bricker NS et al.8
En el estadio III el incremento en amonio fue menor que en los otros estadios debido a que el riñón enfermo casi había alcanzado su máxima capacidad de formar amonio. La FG en ese estadio III era de 14 mL/min comparado con 48 mL/min de ambos riñones en el estadio I. Es decir, una carga ácida en el estadio III no se eliminaría en su totalidad y aparecería acidosis metabólica que ameritaría tratamiento con sales alcalinas.
Producción hormonal en el riñón enfermo
Una de las complicaciones habituales en la insuficiencia renal es la anemia, producto de un déficit de eritropoyetina (EPO), que es una glicoproteína que se genera en los fibroblastos del intersticio renal como se ejemplifica en la Figura 12. Estos fibroblastos están presentes en la parte profunda de la corteza en vecindad con la médula externa. La lesión renal progresiva que afecta el intersticio reduce el número de los fibroblastos productores de esta hormona. En las nefropatías túbulo intersticiales esta lesión es más aparente que en las glomerulopatías.21,22 La excepción es el riñón poliquístico del adulto en que la producción de la EPO está elevada y se acompaña de policitemia; la razón es un aumento en el HIF1α que es el factor inductor de EPO por hipoxia que se acumula alrededor de los quistes.
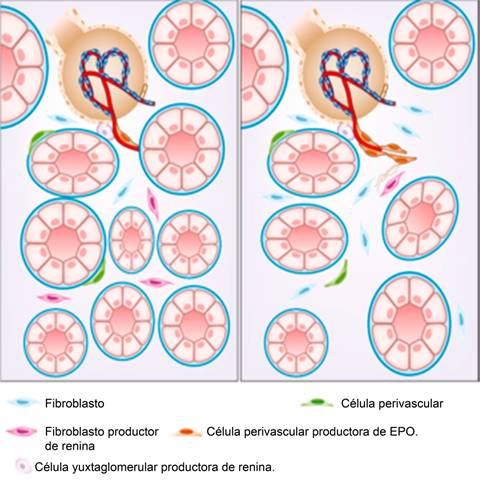
Figura 12: Fisiopatología del intersticio renal. La fibrosis del intersticio es secundaria al daño renal crónico. El esquema ilustra los cambios cualitativos y cuantitativos que definen el cambio de lo fisiológico (izquierda) y los cambios fibróticos del intersticio (derecha). El intersticio no sólo se incrementa en volumen por el aumento en la matriz extracelular, sino también por fibroblastos que generan colágena. Mientras que los fibroblastos productores de EPO se reducen, los fibroblastos perivasculares se incrementan y aumentan la producción de renina. Tomado y modificado de: Zeisberg M et al.22
Otro factor hormonal importante es la renina producida en las células yuxtaglomerulares y fibroblastos perivasculares (Figura 12); este polipéptido precursor del sistema renina-angiotensina-aldosterona es regulador del metabolismo del sodio y de la presión arterial. En la lesión renal túbulo intersticial aumenta su presencia y favorece el estímulo del sistema renina-angiotensina-aldosterona. La matriz extracelular se acumula en el intersticio fibroso. Los fibroblastos productores de EPO se convierten en fibroblastos no productores de EPO. Por el contrario, las células productoras de renina se incrementan a medida que la fibrosis progresa tanto en las células yuxtaglomerulares como en los fibroblastos perivasculares como se observa en la Figura 12.
Un comentario aparte merece la presencia de anemia, que aparece en enfermos renales casi asintomáticos. Con frecuencia pacientes consultan por este problema, y después de la realización de exámenes de laboratorio se encuentran elevaciones de urea y creatinina que indican que la causa de la anemia es daño renal crónico. Por lo que, entre las causas de una anemia normocítica normocrómica de origen no precisado subyace daño renal crónico, particularmente en estadios 3b y 4 de acuerdo a la clasificación de la Guía Kidney Disease: Improving Global Outcomes (KDIGO).23
Conclusiones
Para que el paciente renal se mantenga asintomático con grandes pérdidas de función, el riñon echa mano de esa gran capacidad adaptativa que podemos resumir de la siguiente manera:
1. Las nefronas en el riñón crónicamente enfermo que contribuyen a la formación de la orina lo hacen de una manera organizada e integrada. Existe una clara excepción a este comportamiento en lo que se refiere a la incapacidad para concentrar la orina. Este trastorno es atribuido a defectos estructurales o a adaptaciones funcionales, pero sin duda, el orden funcional como se ha descrito, prevalece en el resto de las funciones del riñón.
2. No se sabe con certeza si algunas nefronas dañadas contribuyen a la función renal, pero si lo hacen es en un porcentaje muy pequeño y no afectan la contribución de las nefronas intactas remanentes que se adaptan progresivamente a la pérdida de más de un millón y medio de nefronas, como ocurre en pacientes con gran daño renal. Estadio 5 de daño renal (FG < 15 mL/min).23
3. La gran discrepancia entre la apariencia estructural del riñón crónicamente enfermo y su función sigue siendo un enigma. Lo que es un hecho que explica esta disparidad es la presencia dentro del parénquima renal enfermo de nefronas muy alteradas y segmentos de nefrona que no contribuyen a la función del riñón. Las nefronas intactas están atrapadas en medio de este daño estructural y en ocasiones su localización es difícil. La microdisección de riñones enfermos es lo que ha permitido observar nefronas de todo tipo: intactas con grandes glomérulos y tubos intactos, nefronas sin glomérulos o con glomérulos alterados y disfuncionales.
4. La información de la literatura de las páginas anteriores permite visualizar un riñón enfermo que conserva sus funciones. Este comportamiento sólo se puede explicar con la hipótesis de la nefrona intacta, que permite al organismo enfermo conservar esa gran estabilidad del medio interno, aun con pérdidas mayores de 80 % de la función renal.
5. Esta destrucción progresiva del riñón propicia la aparición de anemia al destruirse los fibroblastos del túbulo intersticio que sintetizan eritropoyetina. Por otro lado, ocurre un incremento de la renina a expensas de los fibroblastos perivasculares en el túbulo intersticio que estimulan el sistema renina-angiotensina-aldosterona, que propicia la hipertensión arterial, complicación habitual en la enfermedad renal crónica. Es decir, el comportamiento de los fibroblastos del túbulo intersticio es diferente entre los que producen eritropoyetina, que se reducen notoriamente, y los fibroblastos perivasculares, que se incrementan y aumentan la síntesis de renina.

