











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
El mieloma múltiple (MM) es una enfermedad maligna de células plasmáticas de linfocitos B terminalmente diferenciados, que generalmente se asocia con la secreción de inmunoglobulinas monoclonales parciales y/o completas y una constelación de síntomas y signos particulares.1
Se han descrito varias manifestaciones cutáneas asociadas con mieloma múltiple.2 La afección cutánea puede ser resultado de la propia enfermedad o secundaria al tratamiento.3
Bluefarb estimó que aproximadamente el 15% de los pacientes con mieloma múltiple desarrollarían alguna forma de amiloidosis.2
El término amiloidosis fue reportado por primera vez por Rudolph Virchow, médico alemán, en 1854.4 Es un término genérico que significa acumulación extracelular de amiloide, un material homogéneo y acidófilo.5 En la amiloidosis de cadenas ligeras (AL), la proteína precursora de fibrillas amiloides es producida por células plasmáticas monoclonales, presentándose en 5-15% de los pacientes con mieloma múltiple.
La amiloidosis sistémica se caracteriza por el plegamiento incorrecto de las proteínas autólogas, que se agregan en una forma fibrilar anormal y se depositan en los órganos, lo que conduce a una disfunción progresiva.6 Todos los pacientes con amiloidosis sistémica tienen una discrasia subyacente de células plasmáticas.6
En AL, los niveles altos de una cadena ligera de inmunoglobulina monoclonal (Ig) son producidos por una población de células plasmáticas monoclonales, asociada con mieloma múltiple franco o latente en 10 a 20%.7 La amiloidosis de cadenas ligeras lambda es el subtipo más común en amiloidosis cutánea asociada con MM.8 Las alteraciones mucocutáneas están presentes en 20 a 40% de los casos, con frecuencia como los primeros signos de amiloidosis.8
Caso clínico
Paciente femenino de 52 años de edad, con diagnóstico de mieloma múltiple IgA, beta, lambda EC IIA asociado con amiloidosis sistémica, establecido en mayo de 2018. Cuadro clínico inicial con ataque al estado general, fatiga, pérdida de peso e hiporexia.
Inició con dos dermatosis: la primera, localizada en la cabeza, que afectó párpados, región perinasal y mentón, caracterizada por múltiples pápulas céreas lisas, además de afección lingual con macroglosia y festoneado en los bordes laterales (Figura 1); una segunda, localizada en la extremidad superior, que afecta la región axilar, caracterizada por la presencia de una placa color marrón oscuro de superficie anfractuosa acompañada de sensación urente (Figura 2). Recibió manejo quimioterapéutico con ciclofosfamida + bortezomib + dexametasona por seis ciclos, hasta diciembre de 2018, posteriormente, en mayo de 2019, se dio consolidación con trasplante de células progenitoras hematopoyéticas autólogo. Al momento, la paciente se halla en remisión completa.
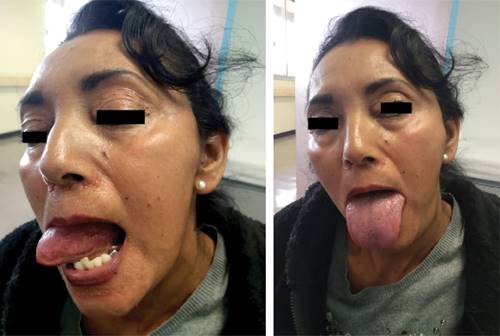
Figura 1: Amiloidosis. Afección en párpados. Afección lingual con macroglosia y borde lateral festoneado, lesiones en regiones perinasales y en mentón.
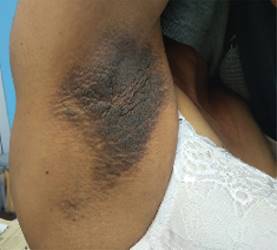
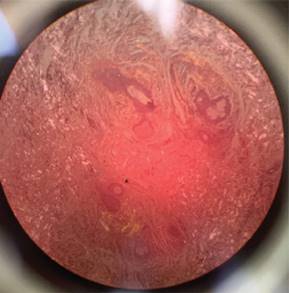
Se le realizó biopsia de la lesión localizada en hueco axilar, en la cual se observó queratosis seborreica reticulada y depósito perianexial focal de material amiloide. Tinción rojo Congo positivo (Figura 3), con lo que se consolida el diagnóstico de amiloidosis cutánea.
Epidemiología
El mieloma múltiple constituye el 10% de todas las neoplasias hematológicas. Fue descrito por primera vez en 1848 y es más frecuente en hombres de la sexta década de la vida.3
La amiloidosis es una enfermedad poco frecuente cuya incidencia exacta se desconoce.9 La prevalencia de amiloidosis asociada con mieloma múltiple varía de 13 a 26% y la presencia de lesiones mucocutáneas en pacientes con amiloidosis asociada con mieloma múltiple varía de 29 a 40%.3
En México, la frecuencia de amiloidosis cutánea en la consulta dermatológica es de aproximadamente 0.5%; predomina en mujeres, con una proporción de 2 a 9:1. Se ha observado a partir de los 14 años de edad y hasta los 72 años, y es más frecuente entre el segundo y quinto decenios de la vida.5
Fisiopatología
No se conoce con exactitud su fisiopatología, sin embargo, se sugieren factores genéticos (10%); los cuales se relacionan con el locus 5p13.1q11.2 y el gen OSMR que codifica para el receptor β de oncostatina M (OSMRβ); éste se expresa en queratinocitos, nerviecillos cutáneos y neuronas nociceptivas.5
La mayoría de los pacientes presentan una forma sistémica de amiloidosis, las más frecuentes son la AL, AA (adquirida) y amiloidosis por transtiretina (ATTR), que es una proteína de transporte tiroidea.7 En la amiloidosis primaria sistémica, también referida como AL, las fibrillas están formadas de inmunoglobulina monoclonal de cadena ligera.5,9
Los niveles elevados sostenidos de SAA (amiloide sérico A), un reactante de fase aguda que forma el amiloide en amiloidosis reactiva o secundaria, son provocados por citocinas proinflamatorias, especialmente la interleucina 6 (IL-6); esto puede ser el resultado de infecciones crónicas, enfermedades reumáticas o autoinflamatorias.7
La característica clave de todos los tipos de amiloidosis es el depósito extracelular de proteínas autólogas, poliméricas e insolubles, en forma de fibrillas anormales, como resultado de la inestabilidad de la estructura terciaria de las proteínas o agregación de proteínas plasmáticas mal plegadas.9
Los depósitos de amiloide consisten en fibrillas finas, constituidas por casi todos los aminoácidos. Existen cantidades variables de IgG, IgM e IgE; componentes del complemento como C1, C3 y C4; fibrinógeno, y proteínas tisulares.5 La infiltración de tejido por estos depósitos de amiloide se produce de forma localizada o sistémica.
Los amiloides se acumulan en la piel, específicamente en las “papilas dérmicas” entre la epidermis y la dermis. Se asocia con fricción crónica o daño epidérmico: amiloidosis por fricción.4
Cuando se localiza, sólo hay afectación cutánea y cuando es sistémica puede ser primaria o secundaria.10 La forma sistémica primaria está relacionada con discrasias ocultas o mieloma múltiple, mientras que la forma secundaria surge de complicaciones de procesos inflamatorios crónicos como artritis reumatoide, osteomielitis, esclerodermia, entre otros.10
Clasificación
Bluefarb clasificó estas lesiones en específicas y no específicas. Las primeras incluyen plasmocitomas extramedulares en la piel o mucosas; las no específicas incluyen aquellas relacionadas con niveles anormales de proteínas (Tabla 1).3
Tabla 1: Manifestaciones cutáneas asociadas con mieloma múltiple.
| • Plasmocitoma cutáneo extramedular. |
| • Amiloidosis cutánea |
| • Pioderma gangrenoso |
| • Vasculitis leucocitoclástica |
| • Escleromixedema |
| • Síndrome de Sweet |
| • Dermatosis subcorneal pustular |
| • Síndrome de POEMS (polineuropatía, organomegalia, endocrinopatía, proteína M y lesiones cutáneas) |
| • Xantomas planos |
| • Hiperqueratosis folicular |
La amiloidosis depende de la extensión del depósito; puede ser sistémica (primaria o consecutiva a otras enfermedades) o cutánea (macular, liquenoide, maculopapular y nodular) (Tabla 2).4),(5
Tabla 2: Clasificación.
| I. Local |
| a) Amiloidosis macular |
| b) Liquen amiloide |
| c) Amiloidosis bifásica o maculopapular |
| d) Amiloidosis nodular o tumefacta |
| e) Amiloidosis discrómica |
| II. Sistémica |
| a) Primaria |
| b) Relacionada con mieloma múltiple |
| c) Secundaria |
| III. Síndromes heredofamiliares |
Modificada de: Arenas R.5
Según la naturaleza bioquímica de las proteínas fibrilares del amiloide, éste se ha clasificado en: A (AA), L (AL), amiloide de proteína de placa central (APCP), senil cardiaco (AScl) y sérico A (SAA).5
Se han descrito tres tipos principales de amiloidosis cutánea primaria: macular (35%), papular/liquen (35%) y mixta/bifásica (15%),11 además de otros tipos más raros.7
La amiloidosis macular se presenta comúnmente en la región interescapular como un parche pigmentado de tamaño variable. Puede ocurrir en otros sitios, como la superficie extensora de los brazos, muslos y espinillas.7 Es típica una apariencia ondulada en sal y pimienta con hiperpigmentación e hipopigmentación alternas.7
La amiloidosis liquenoide es el tipo más común de amiloidosis cutánea.7 Clínicamente, las lesiones aparecen en las regiones pretibiales y antebrazos, como filas lineales de pápulas hiperqueratósicas, agrupadas, pigmentadas y firmes que pueden evolucionar y formas placas grandes.7
Manifestaciones clínicas
La fatiga y pérdida de peso son los síntomas reportados por más de la mitad de los pacientes.12
Todas las formas de amiloidosis sistémica pueden estar asociadas con afectación cutánea oculta o manifiesta.7 Cuando la amiloidosis afecta la piel, puede ser como una manifestación de amiloidosis sistémica, o una amiloidosis cutánea localizada.12
Las enfermedades amiloides cutáneas pueden tener manifestaciones heterogéneas, dependiendo de la ubicación del depósito amiloide dentro de la dermis o la epidermis.7 La afectación de la dermis superior puede causar engrosamiento, aparente como pápulas y placas cerosas, o nódulos.7
Las manifestaciones cutáneas varían, las lesiones comunes incluyen macroglosia y púrpura, las no comunes incluyen cambios esclerodermiformes.3 Las lesiones como petequias, placas purpúricas y equimosis localizadas en los parpados y zonas de flexión, pueden aparecer de manera espontánea o relacionada con traumatismos.3 Las lesiones purpúricas son las más frecuentes y pueden afectar a todo el cuerpo, sin embargo, la lesión característica es la periorbitaria (“ojos de mapache”). Se cree que son secundarias a los depósitos de amiloide en las paredes vasculares, creando fragilidad capilar.8
Las lesiones infiltrativas cutáneas como pápulas, nódulos o placas con aspecto ceroso y hemorrágico pueden localizarse en los párpados, la región retroauricular, el cuello, las axilas, región inguinal y anogenital.3
La presencia de pápulas céreas, placas o nódulos en los pliegues cutáneos y que afectan el área central de la cara son las lesiones más distintivas.8
Una variedad de amiloidosis cutánea se caracteriza también por manchas hipercrómicas, por lo general pruriginosas, permanentes, situadas sobre todo en el tronco.13
Se describen otros tipos de manifestaciones cutáneas, ente ellas la amiloidosis nodular cutánea localizada primaria (PLCNA), también llamada amiloidosis tumefacta, la cual es considerada como la variante más rara de amiloidosis cutánea primaria.14
Manifestaciones linguales. Se presenta en pacientes con mieloma múltiple. Se caracteriza macroglosia y presencia de lesiones pequeñas de color blanco amarillento o blanco grisáceo, asintomáticas; cuando la afección es grande provoca dificultad para la masticación, deglución y fonación.13 La macroglosia es la manifestación más sugestiva y ocurre hasta en el 20% de los pacientes.3
Se pueden encontrar signos dermatológicos en hasta un tercio de los pacientes con amiloide AL.7 Se presenta púrpura periorbitaria en aproximadamente el 10% y las manifestaciones como macroglosia, los nódulos linguales o el surco lateral son exclusivas.7
Cuando ocurre afectación sistémica, los depósitos de amiloide en varios órganos pueden causar afección cardiaca (25-40%) y afección renal (30-50%), que puede progresar a insuficiencia renal grave.3
Diagnóstico
El diagnóstico de amiloidosis se realiza histológicamente mediante la demostración del depósito de amiloide.6 Por lo tanto, es necesaria la realización de una biopsia.12 Rubinow y cols. describieron los hallazgos histopatológicos en un grupo de pacientes con amiloidosis asociada con MM. El hallazgo más común fue la deposición de amiloide en la dermis y tejido celular subcutáneo, además, observaron que el amiloide fue detectado entre los paquetes de colágeno y en algunos casos cerca de la unión dermoepidérmica.12,15
La tinción con rojo Congo, que exhibe una birrefringencia característica bajo luz polarizada cruzada, sigue siendo el estándar de oro.6
Histopatología: en la epidermis se observan hiperqueratosis ortoqueratósica y acantosis, o atrofia, y en la dermis, infiltrado inflamatorio crónico e incontinencia del pigmento.5
En las variedades macular y liquenoide, la sustancia amiloide se origina de los queratinocitos y no se extiende más allá de la dermis papilar.14
Microscopía electrónica: resulta patognomónica e idéntica en todas las variedades; muestra las fibrillas de amiloide como filamentos rígidos no ramificados.5 La determinación del componente de inmunoglobulina involucrado en la amiloidogénesis, resulta de suma importancia, ya que permite guiar la terapia.16
Dermatoscopia: las lesiones papulares muestran un área blanca central rodeada por una gruesa pigmentación marrón a negra. Algunas de las lesiones planas muestran áreas centrales blancas irregulares, rodeadas total o parcialmente por gruesas manchas hiperpigmentadas, mientras que otras lesiones planas sólo muestran manchas irregulares hiperpigmentadas.17
Chuang y cols. Informaron, en un estudio de 35 casos, que en la variedad macular los patrones primarios observados incluían un centro blanco o marrón, rodeado de varias configuraciones de hiperpigmentación y para casos con hiperqueratosis prominente como en la amiloidosis liquenoide, el centro era reemplazado por una área blanca cicatrizal.18
Tratamiento
El tratamiento de la amiloidosis va enfocado a cada tipo.16 Para la forma localizada, se utilizan glucocorticoides tópicos, bajo oclusión e incluso intralesional. Otras modalidades terapéuticas locales incluyen el uso de ácido tricloroacético, podofilina, vitamina A ácida, urea al 40%, ácido retinoico, radioterapia, criocirugía, láser de CO2, neodimio-YAG, luz pulsada, extirpación quirúrgica, apósitos hidrocoloides, luz intermitente intensa, PUVA y UVB de banda angosta.5,11,19 Para los síntomas acompañantes, sobre todo prurito, se recomienda el uso de sedantes o antihistamínicos por vía oral.5
En la amiloidosis AL, que casi siempre es el resultado de una discrasia de células plasmáticas, la terapia antimieloma puede ser efectiva.16,20
Los paradigmas de tratamiento actuales apuntan a suprimir el clon de células plasmáticas para reducir la producción de inmunoglobulinas de la cadena ligera, deteniendo así la deposición de amiloide y permitiendo una respuesta gradual.6
A lo largo del tiempo, se ha recurrido a múltiples terapias farmacológicas, sin embargo, con resultados poco satisfactorios; entre los más utilizados en la forma sistémica se señalan: colchicina, melfalán, glucocorticoides, azatioprina, ciclosporina, acitretina y dimetilsulfóxido.5,19
Actualmente, los regímenes de primera línea para la amiloidosis AL son dosis altas de melfalán y el trasplante autólogo de células progenitoras hematopoyéticas (TCPH autólogo).15 El TCPH autólogo sigue siendo el estándar de atención en pacientes con amiloidosis AL seleccionados.6 Sin embargo, no todos los pacientes son candidatos al procedimiento (20%), por lo que deben ser cuidadosamente investigados para decidir su elegibilidad.21 Los pacientes con un riesgo muy alto incluyen: pacientes > 65 años, con más de dos órganos afectados, aquellos con hipotensión significativa (presión arterial sistólica 90 mmHg) o síncope, tasa de filtración glomerular < 50 mL/min y pacientes con fracción de eyección ventricular izquierda < 45% o espesor de la pared interventricular de 15 mm.21
El bortezomib se ha establecido como el pilar del tratamiento inicial para la mayoría de los pacientes con amiloidosis AL.6 El régimen más utilizado es CyBorD (combinación de ciclofosfamida, bortezomib y dexametasona).6
Venner y asociados informaron una respuesta hematológica hasta en 81.4% de los pacientes que usaban el esquema ECV (ciclofosfamida, bortezomib, dexametasona) de manera quincenal; que es mayor que la respuesta lograda por otras combinaciones, como bortezomib y dexametasona (71%), bortezomib, melfalán y dexametasona (67%) y bortezomib solo (69%).22
Actualmente, se estudia el uso de un anticuerpo monoclonal dirigido al antígeno de células plasmáticas CD38 (daratumumab), el cual se usa habitualmente en el tratamiento de pacientes con mieloma múltiple.21
Kaufman y su equipo describen que la mayoría de los pacientes con amiloidosis AL fallan al cumplir una respuesta terapéutica completa a los esquemas de quimioterapia estándar, y que finalmente los pacientes presentan recaída y progresión de la enfermedad. En su estudio retrospectivo, describen una respuesta hematológica a daratumumab en el 76% de sus pacientes (19 de 25 pacientes previamente tratados con distintos esquemas). Lo que sugiere que la terapia con daratumumab es altamente efectiva para producir una respuesta hematológica en los pacientes con amiloidosis AL refractarios a otros esquemas.23
Pronóstico
El MM generalmente se considera incurable; sin embargo, con los avances terapéuticos recientes, las tasas de supervivencia han aumentado significativamente en los últimos 20 años.1
El pronóstico en los pacientes con amiloidosis con o sin mieloma es pobre, con una media de supervivencia de 12 a 15 meses;3 sin embargo, puede ser tan corta como cuatro a seis meses en pacientes con afección cardiaca e infecciones, siendo las principales causas de mortalidad en estos pacientes.15
Andrei y Wang encontraron que los pacientes con mieloma asociado con amiloidosis AL con afectación de la piel tenían un pronóstico aún peor.15
Conclusiones
El mieloma múltiple raramente afecta la piel;11 15% está asociado con amiloidosis sistémica primaria con alteraciones mucocutáneas como primera manifestación (20-40%), siendo pápulas, placas y nódulos céreos las lesiones más distintivas (pliegues, centrofacial), como en el caso de la paciente. La amiloidosis sistémica y el MM están íntimamente conectados a través de su célula de origen y las manifestaciones cutáneas vistas entre éstos son en ocasiones indistinguibles.15 La asociación de amiloidosis de cadenas ligeras con mieloma múltiple es rara y su tratamiento óptimo no se encuentra bien definido. El trasplante autólogo de células hematopoyéticas es la principal opción terapéutica en pacientes < 70 años con el objetivo de mejorar calidad de vida y contar con periodos libres de tratamiento superiores.6 El pronóstico de la asociación es poco favorable; con una tasa de supervivencia 12-15 meses.3 La paciente citada tiene al momento una SLE desde el diagnóstico de 15 meses y se encuentra en remisión completa.

