











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
La leucemia mielomonocítica crónica1 (LMMC) es un trastorno de células madre hematopoyéticas malignas con características clínicas y patológicas de neoplasia mieloproliferativa (MMP) y síndrome mielodisplásico (SMD). La LMMC se caracteriza por monocitosis de sangre periférica acompañada de displasia de médula ósea comúnmente asociada a citopenias y hepatoesplenomegalia, presenta propensión a progresión de leucemia mieloide aguda (LMA). Históricamente a la LMMC se le ha considerado un subtipo de síndrome mielodisplásico (MDS); sin embargo la LMMC es una entidad clínica y genéticamente distinta; se encuentra entre las leucemias crónicas más agresivas y, con menos terapias efectivas que para la mayoría de las otras neoplasias malignas hematológicas.2 La infiltración extramedular de la LMMC en la piel es poco frecuente; sin embargo, se relaciona en forma directa con mal pronóstico. Al presentarse un paciente en el que se llegó al diagnóstico de LMMC con manifestaciones extramedulares, realizamos la presente comunicación.
Caso clínico
Masculino de 80 años de edad, con diabetes mellitus de 14 años de evolución manejada con hipoglucemiantes orales y dieta, sin otros antecedentes. Relata inicio de padecimiento 60 días atrás al presentar lesiones cutáneas eritematosas diseminadas, manejado por médico general con diagnóstico de alergia, quien prescribió antihistamínicos y esteroides tópicos sin modificación; durante su evolución ha perdido 2 kg de peso, agregándose hiporexia, debilidad muscular e hipertermia ocasional no cuantificada, por lo que acude a urgencias. A la exploración: Paciente en silla de ruedas, con FC de 78 l/min; TA 110/70 mmHg; FR 18 r/min; Temperatura 36.5 oC. Con lesiones dérmicas de tipo nódulo-papulares-eritematosas, algunas confluentes, de bordes bien definidos, algunas de aspecto purpúrico y áreas liqueinificadas por rascado, diseminadas a extremidades, tórax y abdomen (Figura 1), respetando cara, palmas de manos y plantas de pies, en tórax, campos pulmonares libres, ruidos cardiacos rítmicos sin fenómenos agregados; abdomen blando no doloroso, con hepatomegalia de 2 cm y esplenomegalia de 3 cm por debajo de borde costal; peristalsis presente, fuerza muscular disminuida en 3/5 de manera global para las cuatro extremidades; sensibilidad normal, pulsos arteriales periféricos presentes y llenado capilar normal en las cuatro extremidades. Se le hospitaliza y se solicitan exámenes de laboratorio reportando: Bh 9.13 g/dL; CMHG 31.8 g/dL; leucocitos 41,300/mm3; segmentados 52%; bandas 10%; eosinófilos 0%; basófilos 0%; monocitos 27% y linfocitos 8%, plaquetas 40,000/mcL. Al examen de frotis de sangre: metamielocitos 2%; mielocitos 1%; vacuolización citoplasmática de monocitos, basofilia difusa, normoblastos 1% y anisocitosis. Velocidad de sedimentación globular 32 mm/h; PCR 70 mg/L; glucosa 127 mg/dL, urea 81.3 mg/dL; creatinina 2 mg/dL: tiempo de protrombina 17.3 s; tiempo parcial de tromboplastina 30 s; INR 1.27; albúmina 3.4/g/dL; bilirrubina directa 0.7 mg/dL; indirecta 1.5 mg/dL; fosfatasa alcalina 1,283 UI/L; transaminasa glutámico pirúvica 62 UI/L; transaminasa glutámico oxalacética 80 UI/L; gama glutamiltransferasa 310 UI/L. El examen inmunofenotípico en muestra de sangre periférica (Figura 2) mostró: aumento de SS (granularidad) en la población de monocitos, disminución en la expresión de CD4 y HLA-DR, sobreexpresión de CD123 y CD16, además de expresión aberrante de CD56 en 32.8% del total de leucocitos y, asincronismo con freno de diferenciación a nivel de metamielocitos (51.9% del total de leucocitos). La biopsia de piel mostró: piel con infiltración por células neoplásicas malignas dispuestas en nidos, sábanas y/o cordones, con marcador de inmunohistoquímica mieloperoxidasa positivo (Figura 3). Con el diagnóstico de leucemia mielomonocítica crónica se inició tratamiento con hidroxiurea a dosis de 500 mg/cada 12 horas. Se derivó a hematología para control y manejo terapéutico.

Figura 1: Fotografías de lesiones dérmicas de tipo nódulo-papulares-eritematosas, algunas confluentes de bordes bien definidos, algunas de aspecto purpúrico y áreas liqueinificadas por rascado, diseminadas a extremidades, tórax y abdomen, respetando cara, palmas de manos y plantas de pies, en octogenario con diagnóstico de leucemia mielomonocítica crónica.
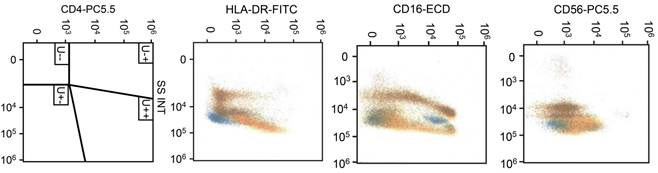
Figura 2: Examen inmunofenotípico en muestra de sangre periférica, mostrando: disminución en la expresión de CD4 y HLA-DR, sobreexpresión de CD123 y CD16, además de expresión aberrante de CD56 en 32.8% del total de leucocitos.
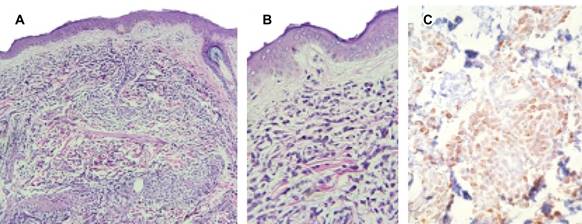
Figura 3: Microfotografías de biopsia de piel: A) Piel con infiltración por células neoplásicas malignas dispuestas en nidos, sábanas y/o cordones ( H&E ). B) Células de citoplasmas escasos, eosinofílicos, núcleos pleomorfos, cromatina en grumos gruesos, mal distribuida con nucléolo presente prominente ( H&E ). C) Marcador de inmunohistoquímica mieloperoxidasa positivo.
Discusión
Respecto a epidemiología, la LMMC se presenta con mayor frecuencia en adultos mayores, con edad promedio entre 65 y 75 años y predominio masculino moderado; se estima que existen alrededor de 1,000 casos diagnosticados anualmente en los Estados Unidos, con tasa de incidencia anual bruta de 0.3 por 100,0003 al igual que en España.
La patogenia de LMMC es poco conocida y compleja, se encuentra una “huella genética” caracterizada por patrones distintivos de genes mutados que originan múltiples clones de células neoplásicas.4 Dentro de las anomalías citogenéticas más comunes se encuentran: a) reordenamientos o deleciones del cromosoma 7 y trisomía 8; b) la médula ósea y las células sanguíneas de la mayoría de los pacientes han adquirido mutaciones en genes que codifican a: modificadores epigenéticos, reguladores de corte y empalme alternativo de mARN, factores de transcripción y señalización de citoquinas; c) gran parte de los casos de LMMC muestran hipersensibilidad al factor estimulante de colonias de macrófagos de granulocitos (GM-CSF) in vitro, lo que probablemente contribuye al fenotipo monocítico de la enfermedad. Ciertas mutaciones genéticas como SRF25 y ASXL1 son más frecuentes, se observan en 50% de los pacientes, siendo su presentación mucho más alta que la frecuencia observada en el SMD.
Las características clínicas de la LMMC no son específicas. Los pacientes pueden acudir al médico por anomalías encontradas en los recuentos sanguíneos de rutina; entre 30 y 50% de los pacientes con leucemia mieloide crónica diagnosticados en Estados Unidos son asintomáticos y por lo general el diagnóstico se realiza por exámenes de laboratorio de rutina;1,2 otros pacientes presentan síntomas o complicaciones como resultado de una citopenia no reconocida previamente, lesiones cutáneas o síntomas relacionados con la esplenomegalia. Clínicamente se debe considerar el diagnóstico de LMCM cuando existe monocitosis periférica persistente e inexplicable en un adulto mayor, en especial en cualquier paciente con diagnóstico patológico de SMD, ya que la monocitosis periférica es el único diferenciador clínico de LMMC en muchos casos. En todos los pacientes, las anormalidades de la sangre periférica y de la médula ósea están presentes a la hora de la presentación.6 La infiltración extramedular del bazo, el hígado, la piel y los ganglios linfáticos es menos común.7
Los primeros casos de infiltración cutánea por LMMC fueron descritos por Duguid8 y colaboradores en 1983; es una presentación infrecuente,9 se considera un signo de mal pronóstico10 siendo además indicador de agudización inminente (transformación blástica) que generalmente tiene lugar en estadios avanzados de la enfermedad,11 como lo demuestra el estudio de Matheus10 y su equipo en 108 casos, en el que 10.2% de los mismos presentaron una supervivencia tan sólo de 7.8 meses después del diagnóstico. Hay algunos casos descritos de infiltración cutánea antes de que se pueda evidenciar patología hematológica en sangre periférica y/o médula ósea (leucemia cutánea aleucémica), aunque la mayoría se desarrolla en el contexto de una enfermedad hematológica ya diagnosticada. El cuadro clínico es muy variable y no existen lesiones cutáneas patognomónicas pudiendo presentarse principalmente como pápulas, nódulos, placas o tumores.10-12 La “leucemia cutis” no es un diagnóstico clínico, se requiere biopsia y estudio histopatológico para su confirmación.13 Se aconseja la combinación de radioterapia y quimioterapia para su tratamiento, ya que la quimioterapia sistémica es adecuada para inducir y mantener en remisión la médula ósea, pero no es eficaz en el control de la infiltración cutánea; inversamente, la radioterapia puede erradicar la enfermedad cutánea, pero no es efectiva en médula ósea.10
En sangre periférica los casos de LMMC presentan monocitosis persistente > 1,000/μL, constituyendo > 10% de toda la diferencial de leucocitos; a pesar de aumento relativo en los monocitos, el recuento total de glóbulos blancos no aumenta en muchos casos de LMMC. La evaluación del frotis de sangre periférica y una biopsia y aspirado de médula ósea son componentes clave para el diagnóstico.1,2 Las condiciones que presentan características similares a LMMC se deben descartar. La inspección cuidadosa del frotis de sangre periférica y el aspirado de médula ósea son necesarios para documentar las características citológicas displásicas identificables en cualquiera o en todos los linajes hematopoyéticos. Dado que el diagnóstico se basa en gran medida en los cambios morfológicos, la calidad de los frotis es de suma importancia. Para determinar el porcentaje de blastos en la sangre periférica se recomienda un diferencial de 200 leucocitos; los frotis de pelaje esponjoso pueden ser necesarios en pacientes con citopenia grave. El porcentaje de blastos en la médula debe calcularse a partir de un recuento diferencial de 500 células realizado en el aspirado de médula ósea. En LMMC, no obstante estar presentes neutropenia, trombocitopenia o coagulopatía, es poco común que se desarrolle una hemorragia o infección en el sitio de la aspiración/biopsia de médula ósea como una complicación del procedimiento.
En cuanto a la médula ósea en LMMC es uniformemente hipercelular,2 con incremento de células mononucleares y sus progenitores, así como células mononucleares anormales que presentan características intermedias entre mielocitos y monocitos. Por otra parte, se observa < 20% de blastos en la médula ósea, incluyendo: mieloblastos, monoblastos y promonocitos; los cuerpos de Auer están ausentes. La fibrosis se puede observar en la médula tanto en pacientes con LMMC como con SMD, su significado aún no está claro.
La evaluación de la médula ósea debe incluir el análisis del cariotipo mediante citogenética convencional;14 si no hay células en división para el análisis de bandas G, se debe realizar una hibridación in situ con fluorescencia (FISH) para excluir el gen de fusión BCR-ABL1 y las translocaciones del cromosoma 5q31-33 que involucran al gen PDGFRB. Se debe considerar un análisis de mutación integral en pacientes con morfología celular no diagnóstica y citogenética normal para establecer la clonalidad. En relación a las anomalías citogenéticas, éstas se identifican en 30% de los casos de LMMC; en el restante 70% no hay marcadores cariotípicos clonales, por lo que el perfil genético de la secuenciación de nueve genes (SRSF2, ASXL1, CBL, EZH2, JAK2, KRAS, NRAS, RUNX1 y TET2) podría identificar un evento clonal en > 90% de los casos sospechosos de LMMC.5 La detección de una mutación somática es evidencia definitiva de clonalidad que, junto con la monocitosis persistente, apoya el diagnóstico de LMMC, además el perfil mutacional está integrado en varias puntuaciones de pronóstico que permiten estratificar los casos de LMMC.15 En ausencia de anomalías genéticas, se debe establecer un diagnóstico de LMMC sólo después de haber seguido al paciente durante semanas o meses con pruebas de laboratorio repetidas en lugar de aplicar el diagnóstico basado en el primer recuento anormal de sangre periférica.
La clasificación de la OMS16 subclasifica los casos en uno de tres grupos, que tienen valor pronóstico: LMMC-0: blastos < 2% en sangre periférica y blastos < 5% en médula ósea; LMMC-1: blastos de 2 a 4% en sangre periférica y/o blastos de 5 a 9% en médula ósea; LMMC-2: blastos de 5 a 19% en sangre periférica, blastos de 10 a 19% en la médula, o presencia de uno o más cuerpos de Auer.
El diagnóstico diferencial1,2 es muy amplio, ya que la LMMC se debe distinguir de otras entidades que también pueden presentar monocitosis o displasia, debiendo diferenciarla de: síndrome mielodisplásico (SMD), leucemia mieloide crónica, mastocitosis sistémica, síndromes mielodisplásicos inclasificables así como causas benignas de monocitosis como el estado asplénico, la sarcoidosis, la enfermedad inflamatoria intestinal) y los trastornos autoinmunes, la depresión mayor y el tratamiento con corticosteroides o factores estimulantes de colonias; por otra parte gran cantidad de infecciones específicas se pueden asociar con monocitosis, entre las cuales están: brucelosis, varicela zoster, endocarditis bacteriana, tuberculosis, malaria, fiebre tifoidea, sífilis y tripanosomiasis; por otra parte existen los pacientes con leucocitosis extrema debida a otras causas como una infección grave o terapia con factores de crecimiento hematopoyéticos, pues pueden tener un recuento absoluto de monocitos > 1,000/μL. Al respecto, el reporte de Selimoglu y colaboradores17 sugiere que muchos de los diagnósticos alternativos podrían descartarse cuando los monocitos CD14 + CD16 representan > 94% del total de monocitos en citometría de flujo. Cuando se utiliza este umbral, la sensibilidad y especificidad para LMMC es de 94 y 92%, respectivamente.
Los agentes de hipometilación, como la 5-azacitidina y la decitabina, se usan comúnmente con tasas de respuesta generales de ~30-40% y tasas de remisión completa de ~7-17% sin impacto en cargas alélicas mutacionales. El trasplante alogénico de células madre es la única opción potencialmente curativa, pero se asocia con una morbilidad y mortalidad significativas.
El pronóstico general de los pacientes con LMMC es malo, con supervivencia media esperada de aproximadamente 30 meses; no obstante, existe heterogeneidad clínica con respecto a la historia natural y la supervivencia, la que se ha abordado mediante al menos nueve sistemas de puntuación de pronóstico distintos, todos ellos utilizan características clínicas y hallazgos de laboratorio, mientras que algunos incorporan análisis citogenéticos; uno de los más utilizados es el modelo propuesto por la Clínica Mayo,18 el cual requiere sólo cuatro variables, disponibles en sangre periférica (recuento absoluto de monocitos, presencia de células mieloides inmaduras, hemoglobina y recuento de plaquetas), de acuerdo al puntaje obtenido se cataloga la supervivencia en mala (10 meses), intermedia (18.5 meses) favorable (32 meses).
Respecto al tratamiento, en la actualidad el trasplante alogénico de células hematopoyéticas (TCH) es la única terapia potencialmente curativa de una proporción pequeña de pacientes con CMML,19 ya que no se ha demostrado que otras opciones modifiquen sustancialmente la historia natural de esta enfermedad. Para aquellos pacientes que no son candidatos a TCH y que deciden no participar en un ensayo clínico, se sugiere terapia dirigida a los síntomas con terapia citorreductora (hidroxiurea)20 o agentes hipometilantes (azacitidina, decitabina).21 Se prefiere la terapia citorreductora para los pacientes con síntomas proliferativos graves, mientras que para los pacientes en los que dominan las citopenias o aquéllos para quienes la hidroxiurea es ineficaz se prefieren los agentes hipometilantes.

