











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkCaso clínico
El presente trabajo aborda el caso de una mujer de 82 años, con antecedentes de importancia de hipotiroidismo en tratamiento, osteoporosis en tratamiento con denosumab, déficit de vitamina D en reposición y fractura por aplastamiento de vertebras torácicas, lo que condiciona a síndrome de cono medular, hernia hiatal corregida por funduplicatura dos años atrás, así como hipertensión de cinco años de evolución en tratamiento con 95 mg/día de metoprolol y 40 mg/día de telmisartán.
Acudió a Urgencias por accesos de tos (antecedente de síncope), con patrón de dificultad respiratoria, náusea y escalofríos de cinco días de evolución. Al ingreso, se le diagnosticó foco neumónico que se corroboró a través de la tomografía computarizada; aunado a esto se observó dilatación esofágica. Por ello, fue tratada con antibiótico como terapia, obteniendo una respuesta satisfactoria de la sintomatología pulmonar; sin embargo, 24 horas después se agregó la incapacidad para eructar, el dolor torácico y la náusea persistente.
Los estudios de laboratorio mostraron hiponatremia (121 mEq/L), hipocaliemia (4.5 mEq/L), hipocalcemia (8.4 mEq/L) e hipomagnesemia (2.3 mEq/L). Aunque se corrigió el desequilibrio hidroelectrolítico tuvo una pobre respuesta al manejo y con altos requerimientos de reposición. La paciente evolucionó con pobre tolerancia a la vía oral e importante distención abdominal, por lo que se decidió iniciar nutrición parenteral (utilizada por cuatro días).
Se efectuó manometría, confirmando acalasia. Se decidió colocar una sonda de gastrostomía y aplicar toxina botulínica en el esfínter esofágico inferior; posterior al evento, se detectó retención aguda de orina, la cual se trató con drenaje por sonda Foley, con uresis inmediata de 800 mL, y de 2,000 mL en el resto del turno. Veinticuatro horas después de la colocación de la sonda de gastrostomía, presentó dolor y distención abdominal importante, así como inestabilidad hemodinámica y estado de choque, por lo que pasó a terapia intensiva; posteriormente, se realizó laparotomía exploratoria, encontrando una hernia interna con dilatación de asas y líquido libre en cavidad, la cual resolvió el problema sin ninguna complicación ni zonas de isquemia mesentérica, mejorando el cuadro de sepsis abdominal.
Durante su estancia en terapia intensiva se reportó, 24 horas posteriores a la laparotomía abdominal, poliuria con uresis de 3,090 mL; para el resto de los días se reportó uresis con promedio de 5,730 mL/día y una variación de 3,000 a 9,000 mL/día. Los balances hídricos se mantuvieron persistentemente negativos (Figura 1), y se presentó normotensa durante su estancia. Se realizó gasometría arterial, con diagnóstico de alcalosis metabólica y alteraciones electrolíticas, hiponatremia, hipocalemia severa, hipomagnesemia e hipocalcemia (Figura 2), que requirió sustitución parenteral con altas dosis de electrolitos (aun así, con pobre respuesta).

Figura 1: Gráficas que revelan uresis (A) y balance hídrico (B) en el paciente con síndrome de Gitelman.
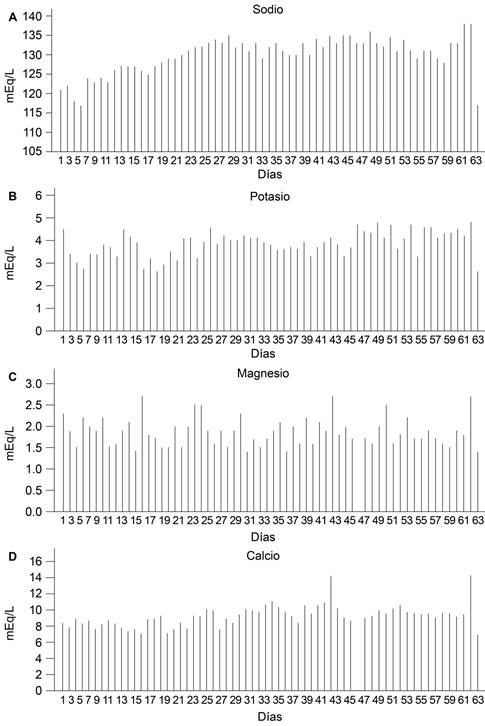
Figura 2: Gráficas que muestran los niveles séricos seriados de electrólitos: sodio (A), potasio (B), magnesio (C), calcio (D), en la paciente portadora de síndrome de Gitelman.
Se utilizó programa de cálculos de función renal (Renal Calc™ Master) (Tabla 1) y se realizó cuantificación de electrolitos urinarios (39 mEq/L; Cl U 131mEq/L; P U 30 mEq/L; K 47.7 mEq/L; Mg U 24.3 mEq/L), que mostraron retención disminuida de los mismos. Por lo anterior, se llegó al diagnóstico de síndrome de Gitelman, entonces se realizaron las correcciones pertinentes del estado hidroelectrolítico y se iniciaron los inhibidores de la COX-2 y ARA II con mejoría del cuadro, por lo que se egresó a terapia intermedia.
Tabla 1: Muestra del uso del programa Renal Calc™ Master (fecha del 17/04/19).
| Entrada 1 | Enfermedad renal crónica, clasificación(ERC, clasif.): estadio 1 (normal) |
| Parámetro (Sp) | Filtración glomerular primaria (FGPEPI) (FGP_EPI): 106.6 mL/min (90-131) |
| Datos del paciente | Creatinina blanco FGP90 (S) (Cr-bfgp90): 0.57 mg/dL (0.5-1.5) |
| Edad: 82 años (0-90) | Creatinina, depuración (Cl Cr) mL/min (90-131) |
| Sexo: mujer | Ácido-base |
| Étnico, factor (ETF) blanco (no-negro) | Brecha aniónica (Suero) (S) (AG): 5.3 mEq/L (8-16) |
| Peso corporal (peso): 60 lb (110-220) | Sodio corregido por glucosa (S) (Na+_c): 127.1 mEq/L (136-145) |
| Estatura (est): 60.6 pulgadas (60-80) | Agua corporal total (TBW): 16.3 L (18-90) |
| H2O porcentaje de PC (% H2O): 60% (50-70) | Agua corporal, déficit (H2O_def): 0 L (0-0) |
| HCO3 volumen de distribución (HCO3 VD): 50% (40-60) | Bicarbonato, déficit (HCO3_def): 0 mEq (0-0) |
| Laboratorio: valores y pruebas | Osmolaridad, calculada (S) (Osm calc): 272.3 mOsm/kg (275-295) |
| Sodio (S) (Na+): 126 mEq/L (136-145) | Brecha osmolar (S) (OsmGap): 0 mOsm/kg (0-10) |
| Potasio (S) (K+): 2.7 mEq/L (3.5-5.5) | Electrolitos |
| Cloro (S) (Cl-): 88 mEq/L (98-106) | Calcio-albúmina, corrección (S) (CaALb_c): 8.3 mg/dL (8.5-10.5) |
| Bicarbonato (S) (HCO3-): 32.70 mEq/L (21-29) | Gradiente transtubular de potasio (TTKG): 7.17% (4-6) |
| Nitrógeno de urea (B) (SUN): 15 mg/dL (10-20) | Potasio urinario, corregido (KU_c): 19.4 mEq/L (25-100) |
| Creatinina (S) (Cr) 0.3 mg/dL (0.5-1.5) | Excreciones fraccionadas |
| Glucosa (S) (gluc): 171 mg/dL (75-115) | Excreción fraccionada de Na+ (FENa): 0.84% (0-1) |
| Albúmina (S) (Alb):2.6 g/dL (3.5-5.5) | Excreción fraccional de urea (FE de urea): 28.23% (20-70) |
| Calcio (S) (Ca++): 7.2 mg/dL (8.5-10.5) | Índice de insuficiencia renal (RFI): 1.06% (0-1) |
| Magnesio (S) (Mg): 2.7 mg/dL (1.8-3) | BUN: Creatinina (S) relación (BUN:CrS): 50 fracción (13-20) |
| Fosfato (S) (PO4): 2.4 mEq/L (1-1.45) | Creatinina (U): creatinina (S) relación (CrU:CrS): 115 fracción (20-40) |
| Osmolaridad (medida) (S) (Osm, m): 266 mOsmol/L(285-295) | UUN:BUN relación (UUN:BUN): 32.5 fracción (3-8) |
| Sodio (U) (Na+): 122 mEq/L (25-150) | Osmolaridad U/osmolaridad S, relación (U:P Osm): 2 fracción (0.5-4) |
| Potasio (U) (K+): 38.2 mEq/L (25-100) | Excreción fraccional de K+ (FEK+): 12.3% (0-100) |
| Nitrógeno de urea, orina (U) (UUN): 487 mg/dL (400-1,200) | Excreción fraccional de magnesio (FEMg): 6.44% (2-4) |
| Creatinina (U) (Cr): 34.5 mg/dL (40-120) | Excreción fraccional de fosfato (FEPi): 10.14% (10-20) |
| Osmolaridad (U) (osm): 525 mOsm/kg (50-1,400) | Peso corporal y masa |
| Volumen (U) (vol): 4.6 mL/min (0.4-1.7) | Índice de Masa Corporal (IMC): 11.5 kg/m2 (18.5-25) 17/4/2019. Bajo Peso |
| Magnesio (U) (Mg): 20 mg/dL (0.97-16.2) | Superficie Corporal Total (SCT): 1.13 m2 (1.3-2.5) |
| Fosfato (U) (PO4): 28 mEq/L (13-28) | Gasto de Energía Basal (GEB): 814.3 kcal/d (1200-25,00) |
| Resultados | Renal Calc™ Master (Print Page). MediCalc® ScyMed |
| Depuraciones | www.scymed.com/es/smnxps/psdcc220_.htm 2/2 |
| Filtración glomerular (Cockcroft) (FGP_cg): 62.2 mL/min (90-131) | |
| Filtración glomerular primaria (MDRD-4) (FGP_mdrd4): 226.7 mL/min (90-131) | |
| Filtración glomerular primaria (MDRD-4-IDMS) (FGP_mdrd4_S): 213 mL/min (90-131) | |
| Filtración glomerular primaria (MDRD-6) (FGP_mdrd6): 169.8 mL/min (90-131) |
La paciente permaneció hospitalizada por 14 días. Al mejorar su estado general, con alimentación por vía oral apoyada por gastrostomía y sin complicaciones, con volumen urinario en las últimas 24 horas de su estancia hospitalaria de 1,932 mL, y electrolitos séricos en rangos normales de Na: 136 mEq/L; K: 4.2 mEq/L; Cl: 99 mEq/L; Mg: 1.8 mEq/L, P: 3.9mEq/L y Ca: 9.6 mEq/L, se decidió su egreso a domicilio con manejo ambulatorio frecuente por nefrología.
Discusión
El síndrome de Gitelman (SG),1 también llamado síndrome de hipocalemia-hipomagnesemia familiar, fue descrito inicialmente en 1966. Éste se caracteriza por una alcalosis metabólica con hipocalemia, asociada con hipomagnesemia significativa y una disminución de la secreción urinaria de calcio. Su prevalencia estimada es de 1 por cada 40,000 casos. Asimismo, es considerada dentro de las enfermedades raras;2 su prevalencia de heterocigotos es de alrededor de 1% en las poblaciones caucásicas, lo que se traduce en una de las enfermedades hereditarias del túbulo renal más frecuentes.
El SG se transmite de forma autosómica recesiva. La mayoría de pacientes presenta mutaciones en el gen SLC12A3 (solute carrier family 12, member 3, por sus siglas en inglés), que codifica para el cotransportador NaCl (NCC)3 sensible a los diuréticos tiazídicos. Actualmente, se han identificado más de 140 mutaciones diferentes a lo largo de toda la proteína. En una pequeña minoría de pacientes, se han identificado mutaciones en el gen CLCNKB, que codifica para el canal de cloro ClC-Kb. El diagnóstico se basa en los síntomas clínicos y en las anomalías bioquímicas (hipocalemia, alcalosis metabólica, hipomagnesemia e hipocalciuria).4
En la mayoría de los casos, los síntomas no aparecen antes de los seis años, y el SG se diagnostica generalmente a lo largo de la adolescencia o en la edad adulta. Con frecuencia, los pacientes presentan periodos transitorios de debilidad muscular y de tetania, a veces acompañados de dolores abdominales, vómitos y fiebre; también son frecuentes las parestesias, especialmente en la cara. Sorprendentemente, algunos pacientes son asintomáticos, exceptuando la aparición, en la edad adulta, de condrocalcinosis que puede manifestarse por inflamación, calor local y dolor en las articulaciones afectadas. La presión sanguínea es, en general, más baja que la de la población general. Ocasionalmente se ha descrito paro cardiorrespiratorio súbito. Comúnmente, en la infancia, el crecimiento es normal, aunque puede retrasarse en infantes con hipocalemia e hipomagnesemia graves.
La sintomatología es variable,5 por ejemplo, el reporte de Dinna y colaboradores,6 realizado en 50 pacientes con diagnóstico establecido de SG con edad X y DE de 40.5 ± 12.5 años y variación de 17 a 74 años (el 70 % perteneciente al género femenino), indica que la prevalencia de los síntomas informados por el paciente en el SG es alta y que éste no es una enfermedad totalmente “asintomática”; la frecuencia de los síntomas es variable, lo cual puede explicarse, en parte, por la discrepancia entre la prevalencia de síntomas informados por el paciente y el médico. Sin embargo, un porcentaje significativo de pacientes califica sus síntomas como de moderados a importantes, enfatizando nuevamente la discrepancia entre la percepción del médico y el paciente, principalmente: fatiga (82%), mareo (80%), síncope (34%), debilidad generalizada (44%), debilidad muscular (70%), calambres (84%), espasmo carpo-pedal/tetania (11%), parálisis (6%), parestesias (62%), artralgia (54%), nicturia (80%), polidipsia (64%), poliuria (50%), sed (76%), enuresis (12%), deseo de ingestión de sal (90 %), hipotensión (62%), vómitos (8%), estreñimiento (16%), dolor abdominal (16%) y palpitaciones (62%).
El síndrome de Bartter es la principal enfermedad genética a tener en cuenta en el diagnóstico diferencial. En este sentido, el consejo genético es importante. La mayoría de pacientes asintomáticos no siguen ningún tratamiento, sólo se les realiza seguimiento que incluye una revisión anual. Se recomienda el uso de suplemento de magnesio a lo largo de toda la vida (óxido de magnesio o sulfato de magnesio). Es necesario proponer un seguimiento cardiológico para detectar cualquier arritmia cardiaca. Asimismo, debe alentarse a los pacientes a que sigan una dieta rica en sodio y potasio. En general, el pronóstico a largo plazo del SG es excelente.

