











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
Covid-19 caught the attention of whole world after its outbreak in the middle of December 2019 from the city Wuhan, China. A few people were diagnosed with pneumonia that was later shown to be due to SARS-CoV-2 [1]. Since then, the epidemic subsequently spread rapidly throughout the world and World Health Organisation declared COVID-19 as pandemic on March11, 2020 [1,2]. Novel coronavirus (2019- nCoV) now designated as SARS-CoV-2 is known to be the causative organism. Third-generation sequencing has revealed that SARS-CoV-2 belongs to the family Coronaviridae, sub family Orthocoronavirinae and order Nidovirales that are positive-sense and single-stranded RNA virus which can be further subdivided into four genera: alpha, beta, gamma, and delta coronavirus [3-5]. SARS-CoV-2 is a beta-corona virus that infects both upper respiratory tract (i.e., sinuses, nose, and throat) and lower respiratory tract (i.e., windpipe and lungs) [6,7]. Patients usually develop symptoms ranging from mild to severe life-threatening, including cough, sore throat, fever, dyspnea, fatigue, diarrhea, acute respiratory distress syndrome, sepsis and septic shock [8]. Furthermore, SARS-CoV-2 infections can cause dysfunctioning or damage of the lungs, heart, brain, kidneys, liver, epidermis, and intestines due to significant increase in the plasma levels of pro-inflammatory cytokines and chemokines, progress to multi-organ failure and death [9-11]. In order to treat current SARS-CoV-2 infection, and more importantly to prepare for unforeseeable new coronaviruses in the future, scientists from the globe response quickly in attempt to identify suitable solutions such as small molecules for the potential therapy or vaccines for the prevention [1]. In such a sensitive situation, expectations from the traditional drug discovery process seem deceptive owing to the time and cost involved. Thus, rapid drug application strategy such as screen of compounds for the inhibition of molecules that have been shown to be essential for attachment, maturation, and replication of SARS-CoV-2 [12]. Recent studies indicate that the following SARS-CoV-2 proteins may be suitable drug development targets: main protease (Mpro) [13], spike glycoprotein -receptor binding domain (SGp-RBD) [14], angiotensin-converting enzyme-2 (ACE2) [15], RNA - dependent RNA polymerase (RdRp) [16], and non-structural protein 15(NSP15, an endonuclease) [17]. Mpro, also known as the 3C-like protease, is required for proteolytic maturation of the coronavirus [18]. This enzyme also plays an essential role in the regulation and cleaving of the polyproteins, pp1a and pp1ab, which generates functional proteins, such as RNA-dependent RNA polymerase, endoribonuclease, and exoribonuclease [19,20]. The inhibition of this enzyme inhibits viral maturation and increases the host immune response to SARS-CoV-2 [21,22]. The spike protein, or S protein of SARS-CoV-2, mediates viral infection and is involved in viral pathogenesis [23]. S1 recognizes and binds to the host receptors namely angiotensin-converting enzyme 2 (ACE2) and subsequent conformational changes in S2 facilitates the fusion of the viral envelope with the host cell membrane [1,24]. The S2 subunit of the S protein has a receptor-binding domain (RBD) that binds to the ACE2 protein [25]. ACE2 is a trans-membrane metalloproteinase that it is a functional receptor for SARS-CoV-2 during the replication cycle [26,27]. The interaction between the viral S protein and the cell membrane receptor (ACE2) causes trimerization of the spike glycoprotein, which allows the internalization of the virus into the host cell to cause infection [28]. Therefore, these two proteins are considered to be the important drug targets for preventing viral infection [29]. The enzyme, RNA-dependent RNA polymerase (RdRp), in combination with the non-structural 7 and 8 proteins, catalyze the synthesis of a positive-stranded RNA molecule that is required for viral translation and replication and thus, infectivity [30,31]. Several studies have shown that the inhibition of the RdRp by compounds such as favipiravir and remdesivir, inhibits the invitro and invivo replication of SARS-CoV-2, thereby significantly decreasing its infectivity [32,33]. Finally, the corona virus uridylate-specific endoribonuclease, NSP-15, has been suggested to be a target for drug development [34], as it mediates replication and processing of sub-genomic RNAs during the replication cycle. It also biodegrades viral polyuridine sequences to decrease the detection of the virus by host cell immune system [35]. It has been reported that natural phytochemicals, such as certain bioflavonoid shown to have effective antiviral action and also have in vitro and in vivo anti-inflammatory and antioxidant efficacy [36-40]. Thus, it is possible that such compounds could have efficacy in the treatment of SARS-CoV-2 by decreasing viral infectivity and replication and / or decreasing the inflammatory response. The identification of naturally derived anti-SARS-CoV-2 compounds can be done by rapidly screening various databases using computational approaches, such as molecular dynamics simulations, molecular docking, drug-likeness prediction and in-silico ADME-T prediction [41,42]. The computational screening approach has been shown to decrease the cost and time required for drug discovery [43]. In this study, we screened 50 flavonoids that were previously reported to have antimicrobial and antioxidant efficacy based on docking score and Molecular Mechanics/Generalized Born Surface Area (MM-GBSA) [44]. The purpose of the virtual screening is to identify compounds that bind with high affinity to the active site of Mpro, SGp-RBD, RdRp, ACE-2, and NSP15 in SARS-CoV-2.
Experimental
Materials and methods
Platform for molecular modeling
The computational analysis, including molecular docking, drug-likeness properties and ADME-T prediction, were performed using the Maestro (12.4) molecular modeling interface (Schrodinger. Inc., New York, USA) [45].
Flavonoid library preparation
A dataset of 50 flavonoid molecules, previously shown to have medicinal efficacy, was prepared by retrieving three dimensional structures from the Pubchem database (www.pubchem.ncbi.nlm.nih.gov), to find compounds with significant binding to the active site of the SARS-CoV-2 proteins, Mpro, SGp-RBD, RdRp, ACE-2 and NSP15. The SDF files of the selected flavonoids were retrieved and prepared using the Ligprep module for geometric optimization and energy minimization using Schrodinger software for the docking studies with selected protein targets.
Protein preparation and grid generation
The X-ray diffraction-based, three-dimensional crystallography structures of the five SARS-CoV-2 protein targets with the PDB.ID codes: Mpro (PDB ID:6W63), SGp-RBD (PDB ID: 7BZ5), ACE2 (PDBID:1R42), RdRp (PDBID:7BTF), NSP15 (PDBID:6WXC), with good resolutions, were retrieved from the PDB databank (www.rcsb.org). These protein structures were further optimized for the docking study using the protein preparation wizard of the Maestro molecular modeling interface (Schrodinger). The missing hydrogen atoms were added to the structure and the hydrogen bond network was optimized using PROPKA at pH 7.0. All water molecules beyond 3 Å and bound ligands were removed, and energy minimization was conducted using the OPLS3e force field. Receptor grid boxes were generated using the “Glide’s Receptor Grid Generation” around the active site residues responsible for the anti-viral activity. While preparing the grid for the target proteins, the size of the outer box of the docking grid was fixed at 20 Å × 20 Å × 20 Å and the inner box dimensions were 10 Å × 10 Å × 10 Å.
Structure-based virtual screening
A structure-based virtual screening method was used to remove compounds based on the docking score. The molecular docking study helps to identify the interactions between the essential amino acid residues of the selected protein and the compounds with low energy conformation. The Grid-based Ligand Docking with Energies (GLIDE) program was used to conduct the in-silico molecular docking experiments [46]. All 50 flavonoids present in the dataset were subjected to the structure-based virtual screening using a high-throughput virtual screening (HTVS) docking protocol and further screening of dataset was performed using standard precision (SP), followed by the extra precision (XP) docking mode [47,48]. Two dimensional visualization of the docked ligand-protein complex was done using the Maestro interface.
Free binding energy (MM-GBSA) analysis
XP- docking provided a significant correlation between pose and score but the free binding energy of the ligand- receptor complex is responsible for the potential therapeutic action. The screened flavonoid molecules were further subjected to binding free energies calculations (MM-GBSA) using the Prime module of Maestro, which incorporates the OPLS3e force field, VSGB solvent model and rotamer search algorithms [49,50]. The binding free energy ΔGbind was calculated using the following equation:
where ΔEMM is the difference in energy between the complex structure and the sum of the energies of the ligand and un-liganded receptor. The Δ𝐺𝑠𝑜𝑙𝑣 is the difference in the GBSA solvation energy of the complex and the sum of the solvation energies for the ligand and un-liganded receptor. The Δ𝐺𝑆𝐴 is the difference in the surface area energy for the complex and the sum of the surface area energies for the ligand and un complexed receptor.
ADME-T and drug-likeness prediction
After screening the flavonoid dataset using the molecular docking study results for the five protein targets of SARS-CoV-2, the absorption, distribution, metabolism, elimination, and toxicity (ADME-T) predictions for the top screened compounds were performed using the Qikprop modules of Maestro [51,52] which predicts the pharmacokinetic properties such as oral absorption,Caco-2 permeability, water-solubility, intestinal absorption, skin permeability, blood-brain barrier permeability, metabolism, excretion, and toxicity [53,54]. The drug-like properties of these compounds were also assessed using Lipinski’s rule of five [55].
Results
Computational screening of the therapeutic targets of SARS-CoV-2
The SDF file of 50 flavonoids, with therapeutic efficacy, were retrieved from the Pub Chem database and were screened based on the five potential therapeutic targets of theSARS-CoV-2, main protease (Mpro), spike glycoprotein-receptor binding domain (SGp-RBD), angiotensin-converting enzyme-2 (ACE2), RNA - dependent RNA polymerase (RdRp), and non-structural protein 15(NSP15). The screening of these compounds was done using molecular docking analysis. All compounds were screened, using the multiple docking filters of HTVS to eliminate false positives (Supplementary Table 1). Based on the docking results, 15 compounds were selected based on a docking score > - 5.5 kcal/mol and glide > -45.00 kcal/mol. These selected compounds were further re-docked using the SP mode of Glide. Subsequently, 07 compounds were obtained that had docking scores > -6.5kcal/mole and a glide energy >-55.00kcal/mol. These compounds were subjected to the XP mode of docking and 4 compounds, with a docking score > -8.5kcal/ mole and a glide energy > -65.00 kcal/mole, were identified. Finally, diosmin was screened based on its drug-like properties, using the ADME-T module. The structure and therapeutic action of the most active flavonoid is shown in Table1. The step wise, structure-based screening protocol is shown in Fig. 1.
Table 1 Structure and efficacy of Diosmin.
| Name of Flavonoid | Compound CID | Structure | Source | Therapeutic Action |
|---|---|---|---|---|
| Diosmin | 5281613 |

|
Teucrium gnaphalodes | Treatment of hemorrhoids, varicose veins, poor circulation in the legs (venous stasis), bleeding (hemorrhage) in the eye or gums and antioxidant. |
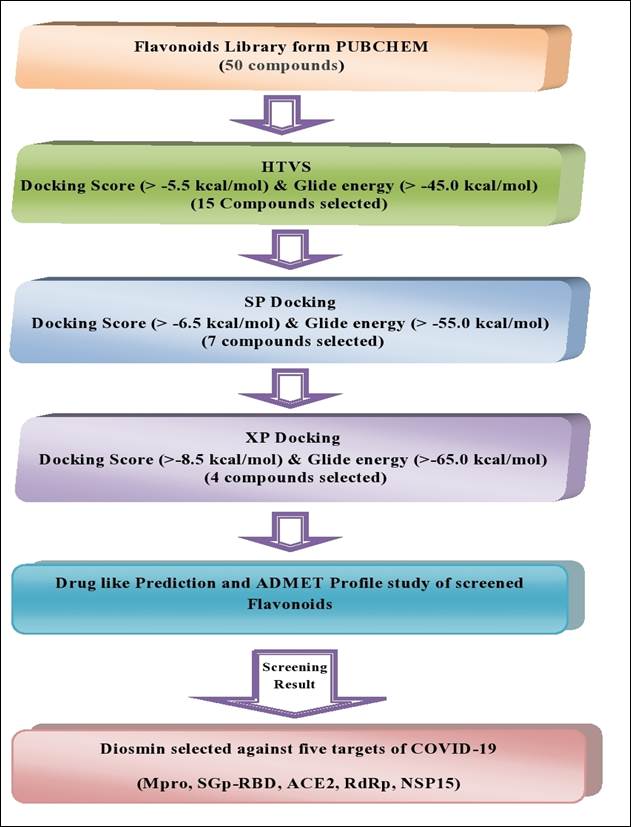
Molecular docking Analysis of diosmin with the active site of five target proteins
The stepwise molecular docking screening protocol HTVS, SP, followed by XP docking mode analysis, were performed to find multi-targeted compounds that had significant docking scores and binding affinity in the grid space of five therapeutically important targets of SARS-CoV-2, Mpro, SGp-RBD, ACE2, RdRp and NSP15. Docking study result reveals that flavonoid, diosmin had docking scores of -12.16, -11.52, -9.75, -11.25, -10.25kcal/mol and glide energy scores of -77.24, -74.49, -65.18, -69.90 and -67.15kcal/mol respectively for five essential drug targets of SARS-CoV-2 (Table 2). The docking scores for diosmin was higher than the re-docking scores of the co-crystallize ligands X77(-6.523 kcal/Mol), remdesivir monophosphate (-7.0 kcal/mol) and tipiracil (-6.05 kcal/mol) for Mpro, RdRp and NSP15 targets of SARS-CoV-2 respectively.
Table 2 Docking scores, Glide energy and Glide e-model energy of diosmin for five targets in SARS-CoV-2.
| Name | Mpro | SGp-RBD | ACE2 | RdRp | NSP15 | ||||||||||
| Dock Score (kcal/ mol) | Glide Energy (kcal/ mol) | Glide e-model energy (kcal/ mol) | Dock Score (kcal/ mol) | Glide Energy (kcal/ mol) | Glide e-model energy (kcal/ mol) | Dock Score (kcal/ mol) | Glide Energy (kcal/ mol) | Glide e-model energy (kcal/ mol) | Dock Score (kcal/ mol) | Glide Energy (kcal/ mol) | Glide e-model energy (kcal/ mol) | Dock Score (kcal/ mol) | Glide Energy (kcal/ mol) | Glide e-model energy (kcal/ mol) | |
| Diosmin | -12.16 | -77.24 | -74.80 | -11.52 | -74.49 | -70.96 | -9.75 | -65.18 | -67.35 | -11.25 | -69.90 | -82.20 | -10.25 | -67.15 | -69.87 |
Interaction of diosmin with the active site aminoacids of the targeted SARS-CoV-2proteins
On the basis of XP dock score diosmin has significant binding poses at the active site of the five potential druggable targets of the SARS-CoV-2 virus. The docking pose and binding interaction between diosmin and different amino acid residues present at the active site of target proteins was presented in Table 3. Diosmin has the best docking interaction with different amino acid residues present at the active site of the Mpro enzyme by forming H-bond, hydrophobic, negative, and positive charge interactions, and polar interaction. The molecular docking results illustrated that the identified ligand diosmin adapted binding modes similar to co-crystallize ligand (X77) within the active site of Mpro, thus confirming the robustness of the docking procedure which interacted with the active site through six H-bonds (His41, Cys44, Asn142, Gly143 and Glu166) and hydrophobic interaction (Pro168, Leu167, Met165, Val42, Cys44,Met49, Leu27, Leu141 and Cys145) (Fig. 2). The high docking scores and significant binding interactions of diosmin occurred with the dyad catalytic site of Mpro.
Table 3 Docking interactions of Diosmin with various amino acid residues at active site of the five targets in SARS-CoV-2.
| Target Protein | H-Bond | Hydrophobic bond | -Ve charge Interaction | +Ve charge interaction | Polar interaction | π -π stacking |
| Mpro | Hie41,Cys4, Asn142, Gly143,Glu166 | Pro168,Leu167,Met165, Val42,Cys44,Met49, Leu27,Leu141,Cys145 | Glu166 | Arg188 | Hie164,Hie163,Gln189,Hie41, Thr45,Ser46,Thr24,Thr25, Asn142,Ser144 | -- |
| SGp-RBD | Arg466,Arg355, Glu516,Phe515, Asp428 | Leu518,Leu517,Phe515, Phe424,Pro426,Tyr396, Trp353,Ile468,Phe464, Pro463 | Glu516,Asp428, Glu465,Asp467 | Arg355,Arg457, Arg466, Lys462 | Ser514,The430 | -- |
| ACE-2 | Arg273, Asn272, Glu375,Glu406, Arg518 | Pro346,Ala153,Ile446, Tyr515,Phe274 | Glu375,Glu402,Glu406 | Arg273, Arg518 | Asn149,Thr371,His374,Ser409, Gln442,Gln522,Thr445,Hie505,Thr276,Asn277 | -- |
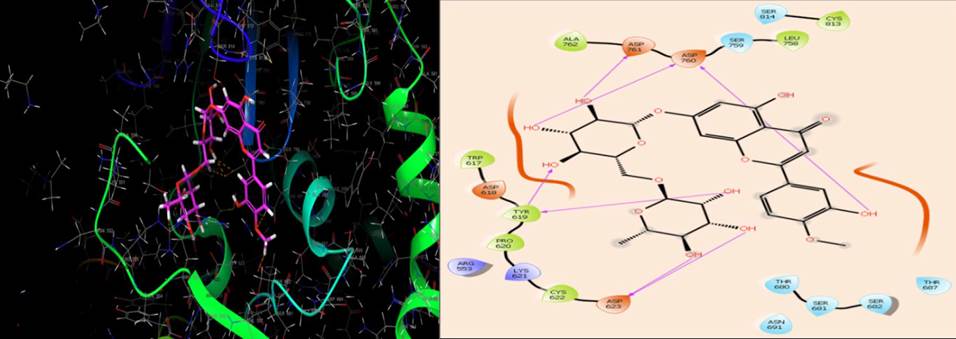
| RdRp | Asp623,Tyr619, Asp760,Asp761 | Cys622,Pro620,Tyr619, Trp617,Ala762,Leu758, Cys813 | Asp623,Asp618, Asp761,Asp760 | Arg553,Lys621 | Thr680,Ser681,Ser682,Thr387, Asn691,Ser759,Ser814 | -- |
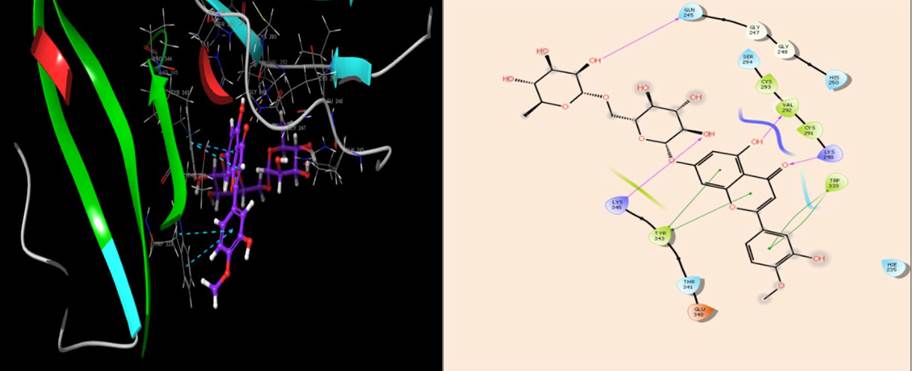
| NSP15 | Lys290,Val292, Gln245,Lys345 | Tyr343,Lys293,Val292, Ys291,Trp333 | Glu340 | Lys345, Lys290 | Hie235,Thr341,Gln245,Ser294, His250 | Tyr343,Trp333 |
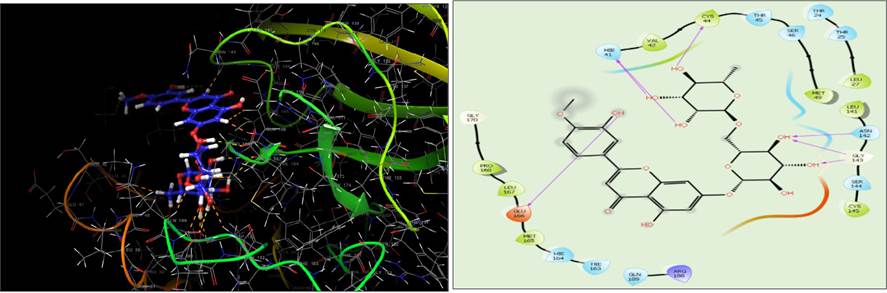
The amino acid residues, 333-527, have been shown to be present at the active site of the binding domain for SGp-RBD [56]. The protein-ligand interaction results indicated that diosmin was bound to the active site and their interaction poses are shown in Fig. 3.The results of the interaction study (Table 3) indicated that diosmin binds to the residues at the active site by forming six H-bond with Arg466, Arg355, Glu516, Phe515, Asp428 and hydrophobic interactions with Leu515, Leu517, Phe515, Phe429, Pro426, Tyr396, Trp353, Ile468, Phe464 and Pro463,hence it is possible that diosmin can inhibit the binding of SGp-RBD to ACE2.
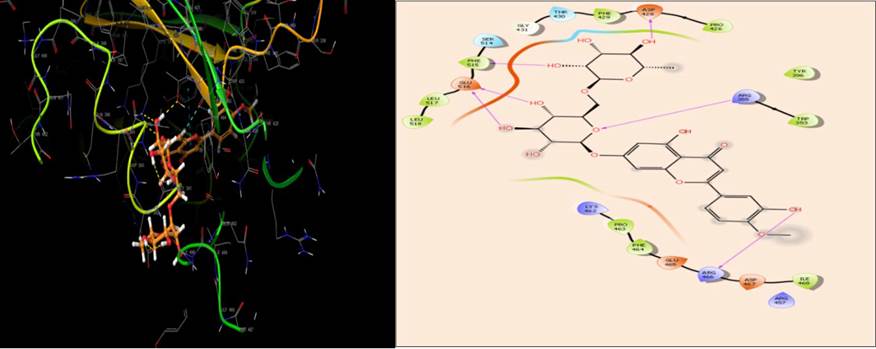
The amino acid residue, Arg273, Gln24, Asp30, His34, Tyr41, Gln42, Met82, Lys353, and Arg357 are involved in the interaction of ACE-2 with binding domain of spike protein of SARS-CoV-2 [56]. The results of the interactions indicated that diosmin forms seven H-bonds with Arg273, Asn277, Glu375, Glu406, Arg518 and hydrophobic interaction with Pro346, Ala153, Ile446, Tyr515 and Phe274, at the active site of the target protein (Fig. 4). Diosmin interacted with Arg273, suggesting that this compound could inhibit the formation of the SGp-ACE2 complex, thereby preventing the entry of SARS-CoV-2 into the host cell.
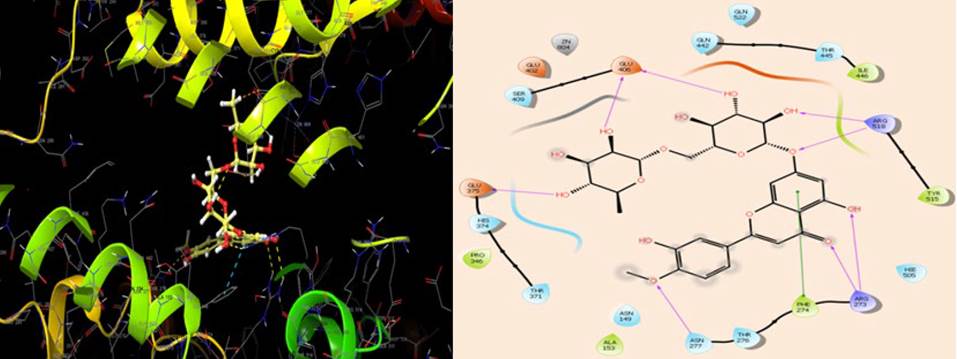
The active site of SARS-CoV-2 RdRp is located in the divalent cationic amino acid 611-626 and catalytic residues in amino acids 753-769[57]. The molecular docking results indicated that diosmin adapted binding modes similar to the RdRp inhibitor, remdesivir monophosphate, at the active site of RdRp [58]. Diosmin formed seven H-bonds (Asp623, Tyr619, Asp760 and Asp761) and hydrophobic interactions (Cys622, Pro620, Tyr619, Trp617, Ala762, Leu758, Cys813) at the active site (Fig. 5). It is possible that diosmin could inhibit SARS-CoV-2 RdRp, thus inhibiting RNA synthesis and replication in the host cell.
The catalytic triad, His235, His250, and Lys290, have been shown to mediate the hydrolytic activity of NSP15 during the replication of SARS-Cov-2 [35]. The results of the docking interaction of diosmin with the active site amino acids of NSP15 indicated that diosmin was interacting with the catalytic triad by forming four H-bonds with Lys290, Val292, Gln245 and Lys345, and hydrophobic interactions with Tyr343, Lys293, Val292, Cys291 andTrp333 (Fig. 6). The results of the interaction pose suggest that diosmin could inhibit the interaction of the ribonucleotides with the catalytic triad of NSP15, which would inhibit SARS-CoV-2 replication and proliferation in the host cell.
ADME-T and drug-likeness prediction study
The use of Lipinski’s rule of five indicated that diosmin has suitable drug-like properties. The results for ADME-T and physiochemical properties of the identified compound are shown in Table 4 and Table 5. The values for molecular weight, Log P and hydrogen bond acceptor potential of diosmin are within the acceptable range. The principal descriptors total solvent accessible surface area (SASA), the hydrophobic component of the SASA (FOSA), hydrophilic component of the SASA (FISA), π (carbon and attached hydrogen component of the SASA (PISA) and percent human oral absorption (POHA), were also with in the range of accepted values. Overall result indicates, diosmin was within the limitations for lipophilicity, hydrophobicity and polarity, suggesting they have drug-like properties.
Table 4 The calculated absorption, distribution, metabolism and excretion of diosmin.
| Name | QPlog Po/w (-2.5 to 6.5) | QPlog S (-6.5 to 0.5) | QPlog HERG (above -5.0) | QPPCaco (< 25% poor, >500 high) | QPlogBB (-3 to 1.2) | QPPMDCK (< 25% poor, >500 high) | QWPlogKp (-8.0 to -0.1) | QPlogKhsa (-1.5 to1.5) |
| Diosmin | -1.226 | -3.047 | -3.861 | 35.58 | -1.137 | 36.54 | -6.158 | -1.106 |
QPlog Po/w: Predicted octanol/water partition coefficient; QPlog S: Predicted aqueous solubility; QPlog HERG:Predicted IC50 value for blockage of HERG K+ channels; QPPCaco: Predicted apparent Caco-2 cell permeability in nm/sec.; QPlogBB: Predicted brain/blood partition coefficient; QPPMDCK: Predicted apparent MDCK cell permeability in nm/sec; QPlogKp: Predicted skin permeability, log Kp; QPlogKhsa: Prediction of binding to human serum albumin
Table 5 Drug likeliness predictions of diosmin.
| Name | Mol. Wt (130-725) | SASA (300-1000) | FOSA (0-750) | FISA (7-330) | PISA (0-450) | Volume (500-2000) | PHOA (< 25% poor, >80% high) |
| Diosmin | 608.552 | 851.625 | 281.55 | 314.004 | 206.071 | 1650.907 | 35.21 |
Mol. Wt: Molecular weight; SASA: Total solvent accessible surface area; FOSA: Hydrophobic component of the SASA; FISA: Hydrophilic component of the SASA; PISA: Pi- carbon and attached hydrogen) component of SASA; PHOA: Percent human oral absorption
Free binding energy (MM-GBSA) calculation
To verify the binding affinities of diosmin, the binding free energy ΔG bind values were calculated by the Prime/MM-GBSA protocol, using the experimental minimum inhibitory concentration (MIC) values. The results of the free binding energy study for diosmin with the five protein targets of SARS-CoV-2 are presented in Table 6.
Table 6 Binding free energy and its individual components in kcal/mol.
| Name | Mpro | SGp-RBD | ACE2 | RdRp | NSP15 | ||||||||||
| MMGBSA dG Bind | MMGBSA dG Bind Coulomb | MMGBSA dG Bind H-bond | MMGBSA dG Bind | MMGBSA dG Bind Coulomb | MMGBSA dG Bind H-bond | MMGBSA dG Bind | MMGBSA dG Bind Coulomb | MMGBSA dG Bind H-bond | MMGBSA dG Bind | MMGBSA dG Bind Coulomb | MMGBSA dG Bind H-bond | MMGBSA dG Bind | MMGBSA dG Bind Coulomb | MMGBSA dG Bind H-bond | |
| Diosmine | -45.29 | -55.56 | -4.42 | -55.6 | -59.74 | -6.54 | -68.24 | -57.25 | -4.86 | -65.92 | -65.96 | -7.21 | -45.03 | -39.82 | -5.12 |
Discussion
Literatures are available for the antiviral efficacy of flavonoids and their derivatives in several viral strains, including avian influenza strain H5N1, HIV, HSV and Ebola [59]. In this study, we used computational approach on a library of 50 flavonoids to detect compounds with high binding affinity to the followingSARS-CoV-2 proteins, main protease (Mpro), spike glycoprotein-receptor binding domain (SGp-RBD), angiotensin-convertingenzyme-2(ACE2), RNA-dependent RNA polymerase (RdRp), and non-structural protein15(NSP15, an endonuclease). The results of docking study, ADME-T and drug likeliness properties ascertain that diosmin has drug-like properties.
The active site of Mpro has been reported to have 4 sub-sites: S1′, S1, S2 and S4. The catalytic Cys-145 and His-163 are located in the S1’ site. The Glu-166 residue is in the S1 pocket, whereas Met-165 and Val-186 are in the S2 pocket and Pro-168 and Gln-189 are located in the S4 site [60]. As compared to other receptor sites in the SARS-Cov-2 proteins, the Mpro site is comparatively flexible and can accommodate a wide variety of ligands, including peptides and small molecules. In the present study, the docking score and glide energy of diosmin within the binding pocket were -12.16 and -77.24 kcal/mol respectively. Interaction of diosmin with the active site through six H-bonds (Hie41, Cys44, Asn142, Gly143 and Glu166) and hydrophobic interaction (Pro168, Leu167, Met165, Val42, Cys44, Met49, Leu27, Leu141and Cys145). Our in-silico results indicate that diosmin had better interactions with Mpro of SAR-CoV-2.
Diosmin interacted with residues at the active site of binding domain for SGp-RBD by forming six H-bond with Arg466, Arg355, Glu516, Phe515, Asp428 and hydrophobic interactions with Leu515, Leu517, Phe515, Phe429, Pro426, Tyr396, Trp353, Ile468, Phe464 and Pro463. The binding score was - 11.52 kcal/mol and a glide energy score of -74.49kcal/mol.
ACE2 has been shown to be largest functional target for SAR-CoV-2. ACE2 is a structural trans membrane receptor for spike protein that is internalized into host cells. The essential residues in ACE2 that are involved in the interaction with the RBD of spike protein include Gln24, Thr27, Lys31, His34, Glu37, Asp38, Tyr41, Gln42, Leu45, Leu79, Met82, Tyr83, Asp90, Gln325, Glu329, Asn330, Lys353, and Gly54. Glu22, Glu23, Lys26, Asp30, Glu35, Glu56, and Glu57 are also involved in the interaction, amino acids, Lys26 and Asp30 are critical for the binding of ACE2 with RBD. Thus, blocking these essential amino acid residues of ACE2 by small molecules or antibodies could prevent the spike protein from binding to the host cell. Docking of diosmin with ACE2 has score - 9.75kcal/mol and binding energy - 65.18 kcal/mol, interacts with ACE2 by forming eight H-bonds with Arg273, Asn272, Glu375, Glu406, Arg518 and hydrophobic interactions with Pro346, Ala153, Ile446, Tyr515 and Phe274aminoacid residues at the active site. The interactions of diosmin could affect Spike RBD binding at this site and future in vitro studies should be done to verify this possibility.
RdRp, in combination with the proteins, Nsp7 and Nsp8, is involved in the replication of the SARS-CoV-2 RNA genome [61].In the present study, the docking score of diosmin for the SARS-CoV-2 RdRp was -11.25 kcal/mol and its glide energy score was -69.90kcal/mol. Seven H-bonds(Asp623,Tyr619,Asp760 and Asp761) and hydrophobic interactions (Cys622, Pro620, Tyr619, Trp617, Ala762, Leu758, Cys813) are formed by the diosmin at the active site as co-crystallized ligand, remdesivir. Thus, diosmin could be a potential inhibitor of SARS-CoV-2RdRp.
Non-structural protein15 (NSP15) plays an essential role in the progression of SARS-CoV-2 infection in the host cells. It is a nidoviral RNA uridylate-specific endoribonuclease that is primarily involved in the evasion of the host immune response. Thus, compounds that potently inhibit NSP15 could have anti-SAR-CoV-2 efficacy. Diosmin had a docking score of -10.25kcal/mol and a glide binding score of - 67.15 kcal/mol, which interacted with the active site of NSP15 by forming four H-bonds with Lys290, Val292, Gln245 and Lys345, and hydrophobic interactions with Tyr343, Lys293, Val292, Cys291 and Trp333 indicate diosmin could be a more potent inhibitor of the NSP15 protein.
The ADME-T results indicated that diosmin had acceptable physicochemical and drug-like properties and within the limitations for lipophilicity, hydrophobicity and polarity. Again, favorable pharmacokinetic properties and no violations to Lipinski’s rule of five indicate diosmin could be used as a potential anti-SAR-CoV- 2 compound. Results of the present study suggest that diosmin interacts effectively at the catalytic site of 5 key proteins in SAR-CoV-2 and had favorable pharmacokinetic properties with no violations to Lipinski’s rule of five. Thus, diosmin could be a potential anti-SAR-CoV-2 compound or important structural features required for the development of novel compounds in the future.
Conclusion
In the present study, diosmin is selected as an optimal compound based on in-silico docking and drug-like property analysis for 50 flavonoids taken from the PubChem database. Diosmin shows significant interactions with five therapeutic targets of SAR-CoV-2: Mpro enzyme, SGp-RBD, ACE2, RdRp and NSP15. Future in vitro and in vivo studies must be done to determine if diosmin is efficacious in SAR-CoV-2.

