











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
Atrazine (ATZ, 2-chloro-4-ethylamino-6-isopropylamino-1,3,5-triazine, C8H14ClN5, M = 215.68 g mol-1) is one of the best known triazine herbicides, extensively used to control the growth of broadleaf weeds in agricultural crops.[1] This herbicide is an environmental contaminant, because it is frequently detected in ground and surface water, as a result of its mobility in soil.[2] It is estimated that people who use groundwater as primary drinking water source are exposed to at least 0.2 µg L-1 of atrazine.[3] Atrazine can be found up to 21 µg L-1 in groundwater, 42 µg L-1 in surface water and 40 µg L-1 in rainfall agricultural areas.[4] Since atrazine has been linked to several human health effects, a maximum contaminant level of 3 µg L-1 was set in 1991, and it has not been changed since then.[5]
Atrazine has been banned in Europe since 2004, but it is still used around the world, including countries like the USA, China, Mexico, Argentina, Brazil and Ecuador.[6] The use of atrazine in South America has increased over time. In Ecuador, for example, this herbicide is applied to corn and sorghum crops with doses varying between 2 and 4 L ha-1, throughout an area of approximately half a million Ha.[7] After atrazine application, it may undergo different fates, including runoff in surface waters or leaching through the ground. The slow biotic processes involved in atrazine biodecomposition in water and deeper soil layers exacerbate the persistence in the environment not only of atrazine itself, but also of its byproducts like hydroxyatrazine, desisopryl atrazine and desethyl atrazine.[8] Several papers have reported the detrimental implications and toxicological effects of atrazine to aquatic microorganisms,[9] amphibians,[4] and fish.[10] It also produces serious health issues, including cancer oncogenes in the reproductive apparatus and babies’ health problems during gestation.[11]
For atrazine degradation, different traditional physicochemical techniques, including adsorption, filtration and biodegradation, have been tested.[12,13] Although these methods can degrade this herbicide, they generate sludge or other types of wastewater that require post-treatment. Recently, advanced oxidation processes (AOPs) have been widely used for wastewater treatment to remove different organic pollutants in synthetic or real effluents. [14] AOPs are based on the in-situ generation of free hydroxyl radicals (•OH), which can react quickly and non-selectively with organic pollutants, yielding CO2, H2O and inorganic ions as final products.[15]
Electrochemical AOPs (EAOPs) have received increasing attention for the elimination of persistent organic pollutants (POPs).[16] Electrochemical oxidation or anodic oxidation (AO) is the most popular EAOP for removing organic pollutants from wastewaters. It consists in the oxidation of pollutants in an electrolytic cell by direct anodic oxidation (direct electron transfer to the anode) and/or indirect reaction with electrogenerated species formed from water discharge at the anode, such as physically adsorbed “active oxygen” (physisorbed hydroxyl radical M(•OH)) or chemisorbed “active oxygen” (oxygen in the lattice of a metal oxide (MO)) at the anode M. The action of these oxidizing species leads to total or partial decontamination, respectively.[17] Table 1 summarizes some important researches about the atrazine degradation via electrooxidation process, in which we highlight the principal parameters that were studied by various authors, such as initial concentration of atrazine, electrodes used, efficiency degradation and current efficiency.
Table 1 Electrooxidation performance in the treatment of Atrazine.
| Electrodes (Anode-cathode) |
% degradation |
Electrolysis time (min) |
J (mA cm -2 ) |
C
0
of ATZ |
% MCE |
% TOC |
References |
|---|---|---|---|---|---|---|---|
| BDD-graphite | 95 | 240 | 2 | 100 ug L-1 | - | 81.3 | 18 |
| Ti/(IrSnO2)-Pt | 100 | 120 | 60 | 20 mg L-1 | - | 35.15 | 19 |
| Ti/(IrRuSnO2) -Pt | 42.35 | ||||||
| BDD-stainless steel | 90 | 360 | 100 | 30 mg L-1 | 20 | 90 | 20 |
| TI/(RuO2) (IrO2) | 98.7 | 120 | 20 | 10 mg L-1 | 21 | ||
| TI/(RuO2) (CeO2) | 82 | - | - | ||||
| TI/(RuO2) (SnO2) | 68 | ||||||
| BDD-carbon filter | 97 | 480 | 40 | 10 mg L-1 | - | 97 | 22 |
| Ti/(IrO2)-Ti | 95 | 60 | 30.7 | 100 ug L-1 | - | - | 23 |
| Ti/(SnO2 )-Ti | 75 | ||||||
| Pt-Pt | 97.5 | 240 | 70 | 0.05 mM | - | 0 | 24 |
| Ti/(SnO2- Sb2O5/PbO5) |
95 | 180 | 40 | 7 * 10-5 mol L-1 | - | - | 25 |
| Ti-Ti | 76 | 360 | 50 | 1810 COD | 39 | - | 26 |
| BDD-Pt | ˃ 90 | 300 | 13.3 | 20 mg L-1 | - | - | 27 |
C0 = initial concentration
Comninellis [28] explained the different behavior of electrodes in AO, considering two limit cases: the so-called “active” and “non-active” anodes. Pt, IrO2 and RuO2 are typical active anodes, whereas PbO2, SnO2 and BDD are considered to be non-active ones. The proposed model considers the initial reaction (1) in both kind of anodes, which corresponds to the oxidation of water molecules leading to the physisorbed M(•OH):
The surface of a non-active anode (like BDD) interacts so weakly with •OH, that it allows the direct reaction of organic compounds with M(•OH) to yield fully-oxidized reaction products, such as CO2 via overall reaction (2) [28]
The BDD anode possesses a very low adsorption of organics in aqueous solutions. It mainly acts as an inert substrate that favors the interchange of electrons. Thus, BDD(•OH) produced from water discharge by reaction (1) is subsequently involved in the oxidation process of organics.[28-30] Furthermore, the overvoltage for O2 evolution and the rate of organic oxidation from physisorbed M(•OH) are closely related to the M-•OH interaction. In general, the weaker the interaction, the faster is the chemical reaction of organics with M(•OH). Since BDD thin films are the best electrodes verifying this behavior, they have been proposed as the most suitable anode for AO. [29] In contrast, Pt is an active anode with a low production of reactive Pt(•OH), because organics interact strongly with Pt surface and most of the above radicals are transformed into the chemisorbed PtO. [30]
This paper presents the results of degradation of ATZ solutions by AO process, aiming to know the relative oxidation power when using Pt or BDD as the anode. Comparative trials were made to study the effect of current density (j) and ATZ concentration on its decay to clarify the role of generated •OH. The evolution of major intermediates in AO-BDD was quantified by high performance liquid chromatography (HPLC).
Experimental
Chemicals
Atrazine was of reagent grade supplied by Sigma-Aldrich. Desethyl atrazine, desethyl desisopropyl atrazine and cyanuric acid were of analytical grade from Aldrich, Panreac and Merck, respectively. The catalyst and background electrolyte were FeSO4·7H2O and Na2SO4 purchased from Karal and JT. Baker, respectively. Solutions were prepared with ultrapure water from a Millipore Milli-Q system (>18 MΩ cm, 25 °C). The solution pH was adjusted to 3.0 with analytical grade H2SO4 supplied by Merck. Solvents and other chemicals were either of analytical or HPLC grade purchased from JT. Baker and Sigma-Aldrich.
Electrolytic systems and analytical procedures
The experiments were carried out in a stirred tank cell at laboratory scale filled with solutions of 175 mL. In all the assays, the solution was vigorously stirred with a magnetic bar at 500 rpm in order to mix the organics and transport them towards/from the electrodes. As electrodes, a Pt plate (99.99% purity from SEMPSA), graphite rod (from Sofacel) and BDD (thin film deposited on conductive Nb plate from MetakemTM) were used. The geometric area of all electrodes was 3 cm2 and the interelectrode gap was of about 2.5 cm. All the trials were made at a constant j provided by a BK Precision 1627A power supply. Solutions with 10 or 40 mg L-1 of ATZ in 0.050 mM Na2SO4 were degraded at pH 3.0 by AO-Pt and AO-BDD at different j values.
The solution pH was determined with a Pent pH-009(1) A pH-meter. Samples withdrawn from electrolyzed solutions were microfiltered with 0.45 (m PTFE filters from Whatman prior to immediate analysis. The decay in atrazine concentration was monitored by reversed-phase HPLC using an Agilent 1260 Infinity LC, fitted with an Agilent Eclipse C18 PAH (250 mm × 4.6 mm, 3 (m particle size) column, and coupled with an Agilent G4212B photodiode array detector set at λ = 223 nm. These analyses were performed by injecting 20 μL aliquots into the LC upon elution of a 55:45 (v/v) H2O:CH3CN mixture at 1 mL min-1 as mobile phase. The chromatograms displayed a peak for atrazine at retention time (t r) of 6.01 min. The degradation efficiency was then determined as follows [31,32]
where ATZo and ATZt are the herbicide concentration at initial time and time t, respectively. Triplicate runs were made for each trial of concentration decay, and average results were reported in figures with errors < ±2%.
For the detection of major aromatic products, the above Agilent LC system fitted with Hypersil ODS column (length = 125 mm, internal diameter = 5 μm) was used. A mixture of 2:98 (v/v) methanol: phosphate buffer solution (pH 6.94) was eluted in an isocratic mode with a constant flow of 1 mL min-1. The detection was done at λ= 223 nm,24] appearing cyanuric acid at t r = 1.77 min, desethyl desisopropyl atrazine at t r =2.12 min and desethyl atrazine at t r = 2.97 min.
Results and Discussion
Degradation of ATZ by anodic oxidation
The comparative oxidation power of AO for ATZ degradation was checked with two anodic materials, namely Pt and BDD, and a graphite cathode. Solutions with 10 or 40 mg L-1 in 0.050 M Na2SO4 at pH 3.0 and 25 ºC were treated at j values between 0.18 and 0.37 A cm-2 for 180 min. In all cases, its pH dropped slightly to a final value close to 2.6-2.7; this behavior is due to the fact that atrazine can generate many acidic compounds of low molecular weight, which decreases the pH. On the other hand, the protons that are formed in the reaction (1) are compensated by the OH- ions that are formed in the cathode, when water is reduced to H2 gas, as expected if recalcitrant short-chain carboxylic acids are generated as final byproducts.[30-345] In fact, if the solution is electrolyzed without atrazine, no change in pH would be observed.
Fig. 1a shows the normalized ATZ abatement for a 10 mg L-1 herbicide solution at different j values by AO-Pt. Higher j increased the degradation rate due to the concomitant acceleration of reaction (1) generating greater quantities of oxidant Pt(•OH). The exponential drop in concentration of these trials was examined as pseudo-order kinetics according to the expression:
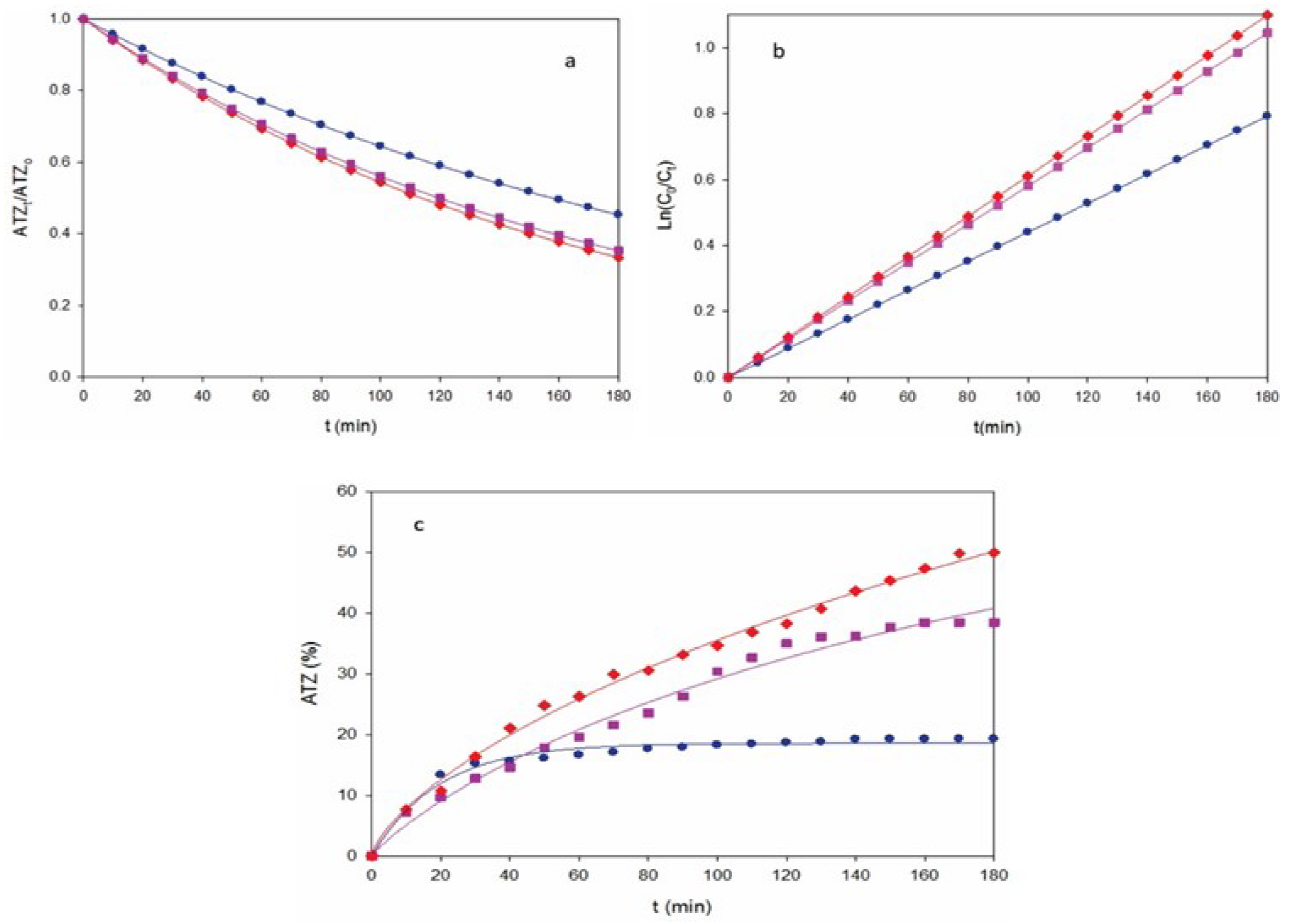
Fig. 1 (a) Normalized atrazine concentration, (b) kinetic analysis assuming a pseudo-first order reaction for ATZ and (c) degradation efficiency with electrolysis time for the AO-Pt treatment of 175 mL of a 10 mg L-1 ATZ and 0.050 mM Na2SO4 solution of pH 3.0 and 25 ºC using a stirred tank reactor equipped with a 3 cm2 Pt anode and a 3 cm2 graphite plate cathode at current density (j): (●) 0.18 A cm-2, (■) 0.27 A cm-2 and (♦) 0.37 A cm-2.
where k 1 is the apparent rate constant. Fig. 1b highlights an excellent fit using Eq. (4), giving rise to increasing k 1-values of 0.0044 min-1 (R 2 = 0.991) for 0.18 A cm-2, 0.0058 min-1 (R 2 = 0.991) for 0.27 A cm-2 and 0.0060 min-1 (R 2 = 0.994) for 0.37 A cm-2. This behavior suggests the formation of a low and steady concentration of Pt(•OH) to attack the ATZ in each j checked.
Fig. 1c presents the change of degradation efficiency with electrolysis time for the assays of Fig. 1a. An enhancement of this parameter can be observed with raising j, finally attaining values of 19%, 38% and 49% for 0.18, 0.27 and 0,37 A cm-2, respectively. These low results indicate on a low ability of Pt anode to remove ATZ molecule. This agrees with the expected low amounts of Pt(•OH) formed at the surface of active Pt, which then possess a small degradation capacity. [30,35]
In contrast, Fig. 2a depicts a much quicker ATZ decay for the same solution by AO-BDD upon comparable conditions, with gradual upgrading of increasing j. This trend can be again accounted for the concomitant acceleration of reaction (1), thereby producing more reactive BDD(•OH) that destroys more rapidly the parent molecule. Fig. 2b illustrates that the corresponding kinetics obeyed the pseudo-first-order reaction of Eq. (4). Increasing k 1-values of 0.0091 min-1 (R 2 = 0.998), 0.0121 min-1 (R 2 = 0.998) and 0.0256 min-1 (R 2 = 0.998) were obtained for 0.18, 0.27 and 0.37 A cm-2, respectively. This tendency corroborates the generation of a low and steady quantity of oxidant BDD(•OH) from reaction (1) in each trial. Furthermore, the degradation efficiency for AO-BDD was much greater than with AO-Pt, as can be inferred from the comparison of Fig. 1c with Fig. 2c. For the former method, 94% of degradation efficiency was reached at 0.18 A cm-2, whereas it was upgraded to high values of 98-99% for 0.27 and 0.37 A cm-2. The degradation superiority of BDD can be related to its ability to generate much greater amounts of BDD(•OH) compared to those of Pt(•OH) formed at the Pt surface. [35]
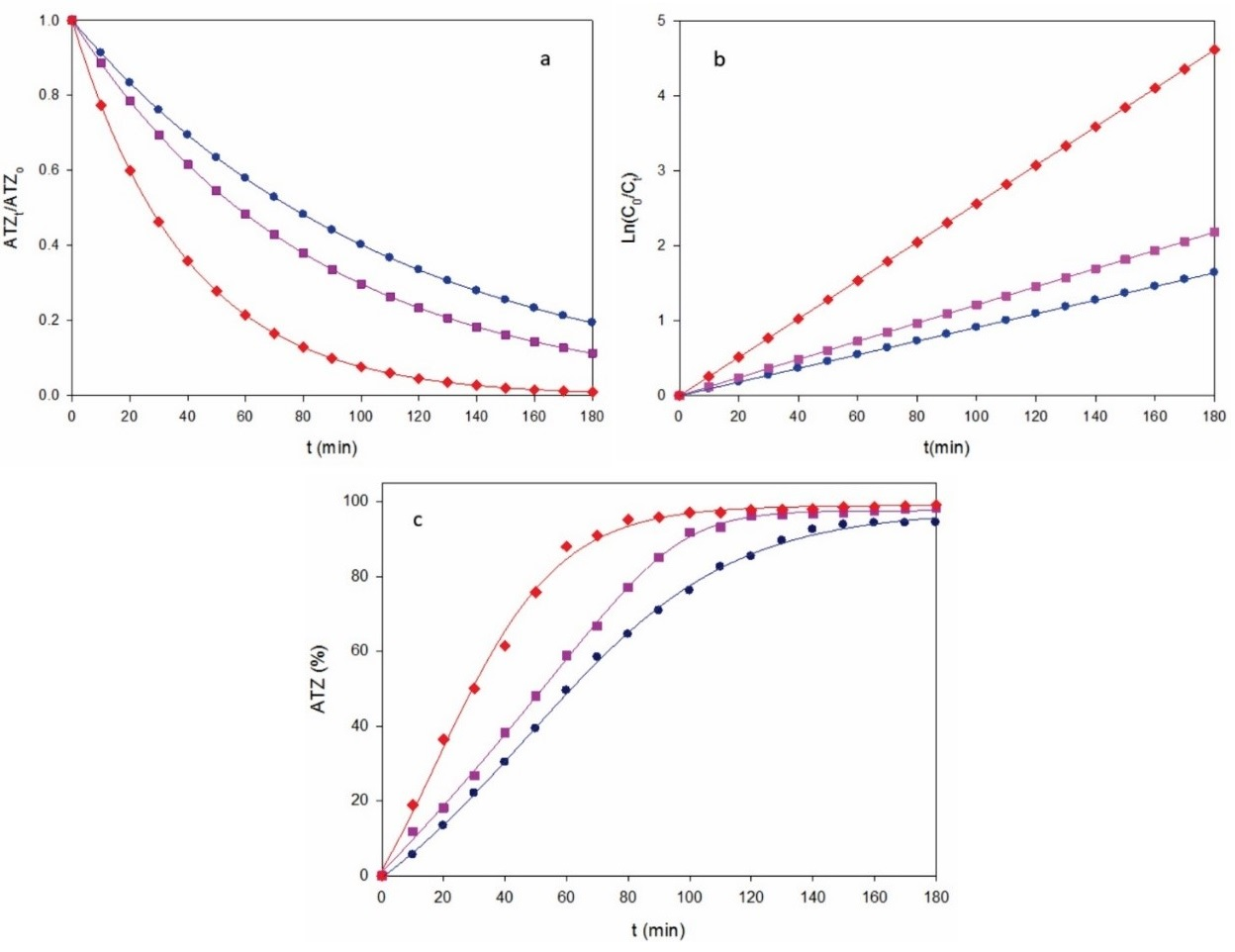
Fig. 2 (a) Normalized atrazine concentration, (b) its kinetic analysis and (c) degradation efficiency vs. electrolysis time for the AO-BDD treatment of the same solution of Fig. 1 using a 3 cm2 BDD anode at j: (●) 0.18 A cm-2, (■) 0.27 A cm-2 and (♦) 0.37 A cm-2.
Fig. 3 and 4 shows the comparative results obtained for the degradation of 40 mg L-1 ATZ, using AO-Pt and AO-BDD, respectively. In all these trials, increasing concentration abatement with growing j was always found, as a result of the enhancement of Pt(•OH) and BDD(•OH) production from the increase in rate of the corresponding reaction (1).

Fig. 3 Time-course of (a) normalized atrazine concentration, (b) pseudo-first-order kinetic analysis of concentration decay and (c) degradation efficiency for the AO-Pt treatment of 175 mL of a 40 mg L-1 ATZ and 0.050 mM Na2SO4 solution of pH 3.0 and 25 ºC using a stirred Pt/graphite tank reactor at j: (●) 0.18 A cm-2, (■) 0.27 A cm-2 and (♦) 0.37 A cm-2.
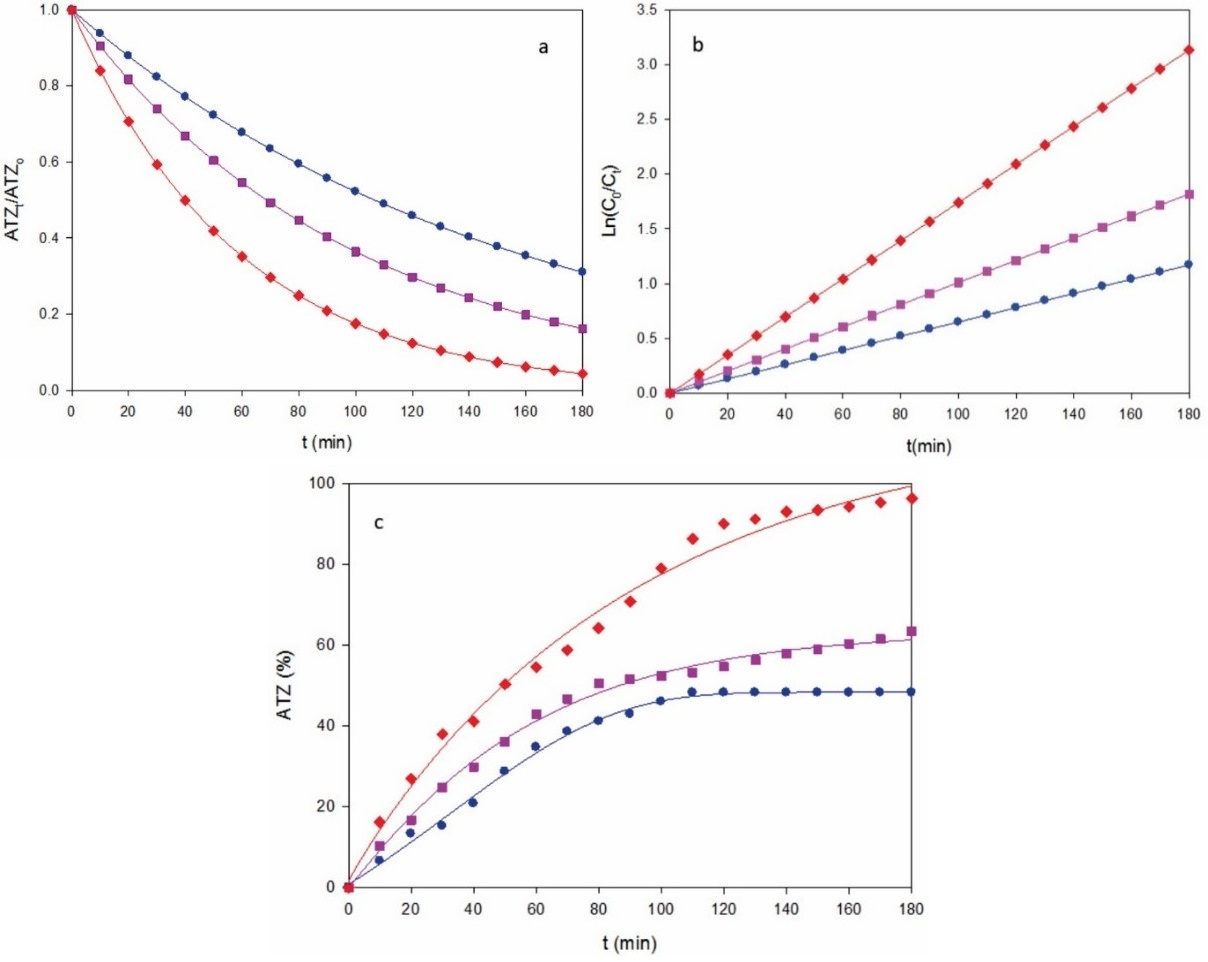
Fig. 4 Change of (a) normalized atrazine concentration, (b) analysis considering a pseudo-first order kinetics for ATZ and (c) degradation efficiency with electrolysis time for the AO-BDD treatment of the solution of Fig. 3 with a stirred BDD/graphite tank reactor at j: (●) 0.18 A cm-2, (■) 0.27 A cm-2 and (♦) 0.37 A cm-2.
The good pseudo-first-order kinetics depicted in Fig. 3b allowed determining k 1-values of 0.0027 min-1 (R 2 = 0.993) for 0.18 A cm-2, 0.0058 min-1 (R 2 = 0.995) for 0.27 A cm-2 and 0.0075 min-1 (R 2 = 0.995) for 0.37 A cm-2. Fig. 3c evidences a rise in degradation efficiency of 39%, 65% and 82% for increasing j values of 0.18, 0.27 and 0.37 A cm-2. Compared to the data obtained for 10 mg L-1 ATZ, lower, equal or higher k 1-values were found at 40 mg L-1, but greater amounts of ATZ were removed in all cases. At j = 0.37 A cm-2, for example, 32.8 mg L-1 of the herbicide were removed for 40 mg L-1, a value much higher than 4.9 mg L-1 determined for 10 mg L-1. Since the same quantity of Pt(•OH) is formed at the same j, more organic events have to occur to enhance ATZ removal, when passing from 10 to 40 mg L-1. This is feasible by the deceleration of parasitic reactions of Pt(•OH), since its reaction with the higher amounts of ATZ and its oxidation products are favored. These parasitic reactions involve, for example, its oxidation to O2 gas, its dimerization to H2O2 and its transformation into the weaker oxidant hydroperoxyl radical (HO2 •) by reactions (5) - (7): [30,34,35]
Similar conclusions can be inferred from the results obtained by AO-BDD. Fig. 4a illustrates the change of herbicide content for 40 mg L-1 ATZ at the j values tested, and Fig. 4b shows the corresponding kinetic analysis. From this behavior, k 1-values of 0.0065 min-1 (R 2 = 0.990) for 0.18 A cm-2, 0.0101 min-1 (R 2 = 0.991) for 0.27 A cm-2 and 0.0174 min-1 (R 2 = 0.993) for 0.37 A cm-2, respectively, which were significantly smaller than those of 10 mg L-1, indicating that k 1 is not a true rate constant, since it depends on the experimental conditions used. The corresponding degradation efficiencies were 48%, 63% and 97% (Fig. 4c), which accounts for the removal of greater amounts of ATZ for 40 than for 10 mg L-1 (Fig. 2c). This is due to the predominant reaction of more BDD(•OH) with higher organic load, which proceeds from the inhibition of its parasitic reactions, similar to reactions (5) - (7).
Regarding the works reviewed in the Table 1, it is possible to observe that several investigations, in which the atrazine electrochemical degradation has been studied, have begun with low concentrations of the contaminant of 100 ug L-1-30 mg L-1, for which they have used 100 minutes of treatment to degrade it, as well as high energy values; they have also tested several electrode materials. However, in our work, we report the results of atrazine degradation in high concentrations of atrazine, 40 mg L-1, which is its solubility limit, and it is also the concentration close to the one which is applied in real crops. Under these conditions, 97% atrazine degradation efficiency is obtained, using only 0.37 A cm-2 of energy and a BDD electrode. In addition, the aromatic products detected are in very low concentration and are not dangerous for the environment. In the light of these results, we are reporting the adequate conditions to carry out a viable treatment of this compound.
Evolution of primary aromatic products
Fig. 5a-c (b) highlights the evolution of the concentration of major primary products, such as cyanuric acid, desethyl desisopropyl atrazine and desethyl atrazine determined by HPLC during the AO-BDD treatments of the 40 mg L-1 ATZ upon the conditions of Fig. 4a. These products proceed from the hydroxylation and/or dealkylation of the parent molecule upon the attack of BDD(•OH). As can be seen, the maximum accumulation of all compounds appears between 120 and 140 min, regardless of the applied j, meaning that at such time a significant amount of generated BDD(•OH) began already to attack the primary products formed. Desethyl atrazine was the product more easily formed, with a maximum of about 80 μg L-1 at 0.37 A cm-2, a value that progressively decreased to about 40 and 12 μg L-1 for 0.27 and 0.18 A cm-2, respectively (Fig. 5b). This suggests a much larger reactivity of atrazine with the higher amounts of BDD(•OH) formed as increasing j to yield desethyl atrazine. A higher rise in cyanuric acid concentration can also be observed in Fig. 5a from 0.18 to 0.27 A cm-2 by the larger BDD(•OH) productivity, which is inhibited by increasing j up to 0.37 A cm-2. This trend was much less relevant for desethyl desisopropyl atrazine, since it presented its maximal value between about 8 μg L-1 at 0.18 A cm-2 and near 10 μg L-1 at 0.37 A cm-2 (Fig. 5c). These results point to the raising initial generation of products in the order: desethyl desisopropyl atrazine < cyanuric acid < desethyl atrazine [18].
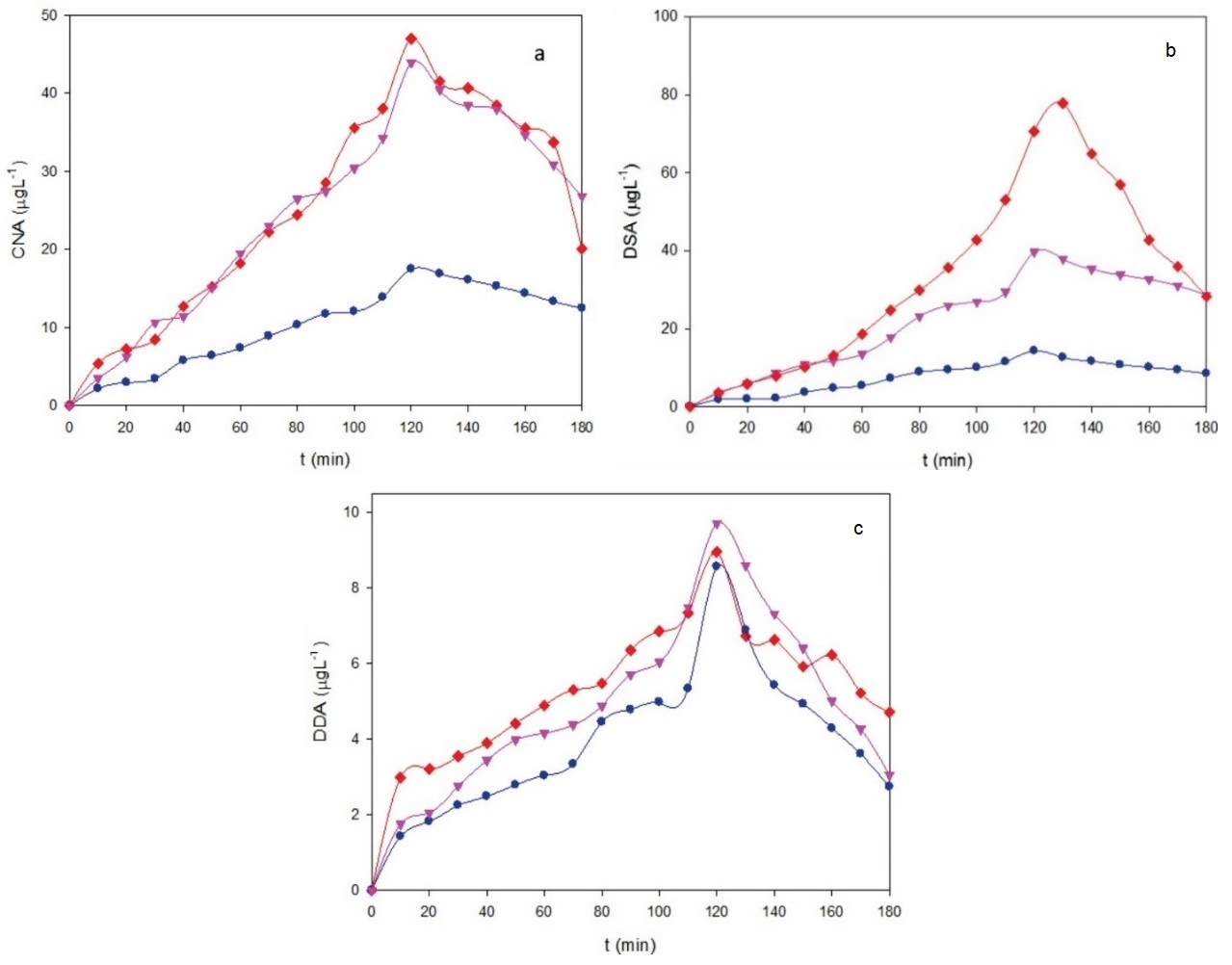
Fig. 5 Time course of the concentration of (a) cyanuric acid, (b) desethyl desisopropyl atrazine and (c) desethyl atrazine during the AO-BDD treatment of Fig. 4 at j: (●) 0.18 A cm-2, (▼) 0.27 A cm-2 and (♦) 0.37 A cm-2.
Conclusions
It has been shown that ATZ in acidic sulfate solution is more rapidly degraded by AO using a non-active BDD anode than an active Pt one. This is due to the higher production of oxidant BDD(•OH) at the former anode. The ATZ decay always obeyed a pseudo-first-order kinetics. Increasing j always accelerated the degradation process by the greater rate of generated hydroxyl radicals, giving rise to the growth of k 1. The rise in ATZ concentration favored its removal due to the concomitant inhibition of parasitic reactions of hydroxyl radicals. The major oxidation products were more largely formed in the sequence: desethyl desisopropyl atrazine < cyanuric acid < desethyl atrazine.

