











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
Oxidative DNA damage has been implicated in aging, diseases and environmental toxicity [1,2]. Oxidative damage to DNA can be produced by reactive oxygen species (ROS), including superoxide radical anion, singlet oxygen, hydrogen peroxide and hydroxyl radical, which are present in the cell as a consequence of endogenous reactions, or from exogenous sources, including ionizing radiation [3,4]. If reactions occur on the nucleobase, the preferential site is guanine, owing to its lowest redox potential (-1.3 V vs NHE) among the four DNA bases [5]. An important event that stems from the secondary oxidation of guanine is the formation of DNA-Protein crosslinks (DPCs). DPCs are extremely bulky lesions that are likely to impair various DNA processes including replication, transcription and repair [6,7]. It has been estimated that the endogenous level of DPCs in human white blood cell range between 0.5 to 4.5 in 107 bases [8] with elevated levels linked to oxidative conditions such as exposure to heavy metals such as nickel and chromium [9-13], which are known carcinogens, and are also shown to be the major lesion associated with arsenite exposure [14]. This supports the theory that DNA-protein cross-link formation is linked to guanine oxidation. Previous work from our group and others, have demonstrated that in vitro, bound proteins readily form cross-links to DNA, likely forming from the secondary oxidation of 8OdG followed by addition of a protein’s nucleophilic amino acid side chain, then forming a hydantoin-based lesion, the identity of which is dependent on the type of reactive oxygen involved [15-23]. For instance, cross-linking in the presence of copper-generated reactive oxygen produces adducts with guanidinohydantoin Gh [18], while singlet oxygen induced oxidative conditions generates spiroiminodihydantoin (Sp) -based adducts [17,19]. Most of the previous reports were conducted on small molecule model systems involving the nucleoside dG and a nucleophilic amino acid such as lysine and tyrosine, given that these models are relatively easy to control and can be further analyzed [15,16,24]. A complete characterization of intact DNA-protein cross-links have been challenging due to adduct instability and multiple oxidative events that can occur in the protein. As a result, DNA-protein cross-links remain as one of the least understood types of DNA damage. The goal of this study is to develop a method to completely characterize DNA-protein cross-links plus identify the site of cross-linking and the type of lesion involved with the aid of ICP-MS/MS via sulfur and phosphorus detection at ultra-trace levels.
To date, many different analytical methods have been used to investigate the cross-linking reaction mechanism and to obtain the chemical structure of DPCs [24,25]. Direct MS analysis of DPCs is challenging because of the different ionization properties of DNA and proteins. Additionally, an appropriate matrix for MALDI (matrix-assisted laser desorption/ionization MS) analysis of DPCs is not yet available. More recently, peptide sequencing and mass spectrometry (MS) as analytical techniques are playing increasingly important roles for the structure determination of DPCs [26]. In our previous study, a novel approach was presented for the sub-ppb detection and quantification of DPCs by newly developed inductively coupled plasma mass spectrometry/mass spectrometry (ICPMS/MS) [27]. This method is an effective technique for purifying DPCs. DNA-protein cross-links were readily isolated and purified via fraction collection of species containing both a sulfur and phosphorus signal from a HPLC-ICPMS/MS system, which is indicative of a molecule containing both protein and DNA and is likely a DNA-protein cross-link.
We aim here to enhance our developed method to allow complete characterization of DNA-protein cross-links. First, a small molecule model was utilized to identify the adduct structure that will likely occur in an intact DNA-protein cross-link. We investigate the thermal stability of DNA-protein cross-links, both in an intact DPC and a small molecule adduct to determine feasibility of digestion/thermal degradation of DNA without cross-link information being lost. Then, thermal degradation was conducted to reduce the cross-linked DNA into a single nucleoside. The remaining protein-nucleoside adduct is proteolytically digested generating a peptide-nucleoside adduct, which in the absence of the phosphate moiety allows for facile structural characterization via electrospray ionization mass spectrometric (ESI-MS) analysis. Additional calculation was done for peptide matching allowing us to determine the location of the cross-link in the protein. Importantly, using peptide MS/MS analysis, we were able to locate the specific amino acid position involved. Additionally, we show that steric effect places an important role in DNA-protein cross-link formation.
Experimental
Materials
All experiments were done in triplicate or greater. All chemicals were purchased from Fisher Scientific (Pittsburgh, PA, USA) unless otherwise noted. The 27-nucleotide DNA, 5’-GGGGCCCCGTCGTTTTACAACGTCGTG-3’, and its complement 5’- CACGACGTTGTAAAACGACGGGGCCCC-3’ were purchased from Eurofins Genomics Operon (Huntsville, AL, USA). Ribonuclease A, Rose Bengal, 2’-Deoxyguanosine, and N-acetyl-Lysine-Methyl ester were obtained from Sigma Aldrich (St. Louis, MO, USA). The sequence-grade modified trypsin was purchased from Promega (Madison, WI,USA).
Methods
2’-Deoxyguanosine-Lys derivative Oxidative Cross-Linking. Cross-linking experiments were carried out by mixing 2’-Deoxyguanosine (dG) and alpha-N-acetyl-Lysine-Methyl ester (AcLysOMe) with photo oxidant Rose Bengal [500 µL; 25 mM sodium phosphate, 4.5 mM 2’-Deoxyguanosine, 9 mM N-acetyl-Lysine-Methyl ester and 0.45 mM Rose Bengal, pH8 in a 1.5 mL centrifuge tube with the lid open and irradiated with an LED lamp providing 3 mW of radiative flux at 375nm. At these conditions, sufficient reaction yields for analysis were observed, and are easily translatable to future cell experiments without major toxicity. During the irradiation, the solution was held at a distance of 3 cm from the radiation source for 4 hours.
Purification of the dG- Ac Lys OMe adduct by RP-HPLC. Purification of the cross-linking adduct from the reaction of 2’-deoxyguanosine and alpha-N-acetyl-lysine-methyl ester was carried as previously reported [27]. A high performance liquid chromatography system (HPLC) consisted on an Agilent 1100 series (Agilent Technologies, Santa Clara, CA, USA), equipped with a binary HPLC pump, an autosampler, a vacuum degasser system, a temperature column compartment, and a diode array detector. The UV absorbance was recorded at from 210nm to 400nm. A Beckman Coulter Ultrasphere C18 [5µm, 4.6mm x 15cm] revered phase (RP) column was used. The column was pre-equilibrated with water containing 0.1% formic acid for 20 minutes before the samples were injected. To identify different peaks on the chromatogram after the cross-linking reaction, the mixture containing all components of the reaction and the controls, which are the two reactants, were injected separately into the system with an injection volume of 2 µL, and the chromatograms overlapped as seen in Fig. 1-A. The compleate parameters for this RP-HPLC separation are given in Table. 1. The dG-AcLysOMe adduct was eluted between 5.0 minute and 5.6 minute, as shown by the increase on intensity with longer reaction times, Fig. 1-B. This fraction was collected and concentrated to 100 µL by freeze-drying for further analysis.
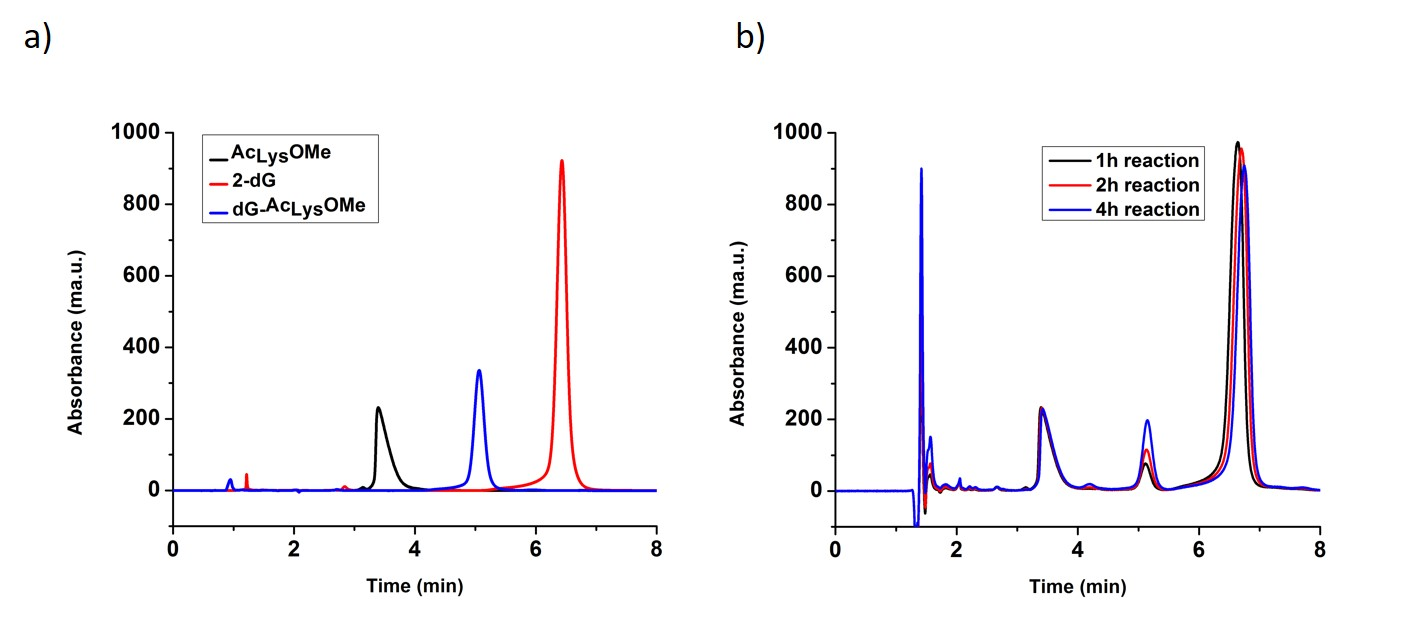
Fig. 1 a) Reversed Phase Chromatogram with UV detection of two reactants (AcLysOMe, 2-dG) and and purified dG-AcLysOMe b) Reversed Phase chromatogram with UV detection of the reaction mix after 1 hour, 2 h and 4 h reaction times
Table 1 Operating conditions for HPLC-CHIP-ESI-ITMS
| HPLC- Chip-ESI- ITMS parameters |
Column | Large Capacity Chip II Zorbax 300SB-C18 (150mm x 75um, 5µm) |
| Flow rate (µL/min) | 3 for capillary flow pump; 0.3 for nano flow pump |
|
| Mobile phases | A, 0.1% formic acid in water; B, 0.1 % formic acid in acetonitrile/water (90/10, v/v) mixture |
|
| Gradient | 0 min 3% B 3 min 3% B 70 min 70% B 75 min 100% B 77 min 100% B 80 min 3% B |
|
| Nano spray needle voltage (V) | 1850 | |
| Injection volume (µL) | 2 | |
| Drying temperature (°C) | 300 | |
| Drying gas (L/min N2) | 3.0 | |
| m/z range | 50-2200 |
Structure determination of the dG- Ac Lys OMe adduct by ESI-ITMS. The identity verification of the dG-AcLysOMe adduct was performed on a 6340 series MSD Ion Trap XCT Ultra system with direct infusion via a syringe pump at a flow rate of 18 µL/h (Agilent Technologies, Santa Clara, CA) with the micro fluidic nanospray chip cube interface, which is automatically loaded and positioned into the MS nanospray chamber. The system was operated in ultra-scan positive ion mode with the maximum accumulation time of 300 msec. The ESI-ITMS was tuned under positive ion mode with the ESI Tuning Mix for Ion Trap (Agilent Technologies, Santa Clara, CA) containing compounds at the following m/z: 118, 322, 622, 922, 1,522, and 2,122. All MSn experiments were performed with an isolation width of 1.2 mass units and the fragmentation amplitude was 0.6 (arbitrary units). Four parent ions were selected and were isolated and fragmented per duty cycle, producing MS2 and MS3 spectra by collision induced dissociation with He gas equivalent to ≈15 eV. Full scan mass spectra were acquired over the m/z range 50-2100 in the positive ion mode. For MS/MS experiments, experimental conditions consisted of: m/z range: 50-2000; isolation width, 2 m/z units; fragmentation energy, 30-200%; fragmentation time, 40 ms.
Study of the thermal stability of the cross-linking covalent bond. The thermal stability study of the cross-linking bond was done with a voltage-controlled heater using an oil bath. Thermal de-composition was used instead of enzymatic hydrolysis for two reasons, simplicity to achieve the desired level of cleavage and to avoid the possibility of steric impediments given the bulky nature of DPCs. A 50 µL volume of the samples from the RP-HPLC peak containing the dG-AcLysOMe adduct were heated to 170 ºC for 1 hour and then reconstituted in 50 µL of water. The product after heating was also analyzed by the ESI-ITMS under the conditions given above.
DNA-Ribonuclease-A oxidative cross-linking and purification. DPCs were synthesized and purified with same methods mentioned in our previous study [38]. In short, the reaction mixture was separated by size exclusion chromatography, where the eluent was directed to an ICP-MS/MS system for phosphorous and sulfur analysis. The presence of phosphorous confirmed the elution profile of the DNA, sulfur of the protein moiety while both at a large molecular mass region was selected as the formed DPC.
Thermal degradation of DNA-ribonuclease-A cross-links and purification of dG-ribonuclease. The mixture containing the purified DNA-protein cross-link was heated to 170 ºC for 1 hour and then re-dissolved in 100 µL of water. Size Exclusion Chromatography (SEC) was used to separate the components into molecular weight fractions following thermal hydrolysis. The column was calibrated using a bio-rad SEC standard containing Thyroglobulin (bovine, 670kDa), γ-globulin (bovine, 158 kDa), Ovalbumin (chicken, 44 kDa), Myoglobin (horse, 17 kDa) and Vitamin B (1.35 kDa).
Trypsin proteolysis of the dG-Ribonuclease A. After the purification of dG-RNase A from the components post-thermal hydrolysis, trypsin was added to the dG-RNAse A adduct to digest the RNAse A portion of the adduct into its peptide components. Water was used as blank and 1 mg/mL Bovine Serum Albumin (BSA) was used as standard to evaluate the extent of proteolysis. Digestion was conducted according to vendor instructions, the reaction was carried out in a pH buffered solution consisting on 15 µL of 50 mM ammonium bicarbonate, 1.5 µL of 100 mM DDT, and 10 µL of sample for digestion in a 0.5 mL tube. The mixture was adjusted to 27 µL with ultrapure water and the sample was incubated at 95oC for 5 minutes. After cooling, a 3 µL volume of 100 mM iodoacetamide was then added into the mixture and incubated in the dark at room temperature for 20 min, followed by addition of 2 µL of sequence grade modified trypsin to a final trypsin concentration of 0.1 mg/mL. The mixture was subsequently incubated overnight at 37oC. 1 µL volume of 88% formic acid was added to the mixture to stop the reaction. The mixture was then subjected to ultrafiltration in order to eliminate trypsin and larger digestion fragments by loading into a 0.5 mL MWCO spin filter with a 10,000 Da (nominal) cut-off membrane and centrifuged for 10 minutes at 10,000 x g. The filtrate containing the peptide fragments was injected into the HPLC-Chip-ESI-ITMS system.
Peptide separation and identification by HPLC-Chip-ESI-ITMS. LC-MS/MS analysis of the peptide mixture was performed on an Agilent 6340 series HPLC- Chip-IonTrap XCT system (Agilent Technologies, Santa Clara, CA), equipped with a Chip Cube interface with an electrospray ionization source, a well plate sampler, a nano- flow binary pump, and a capillary-flow binary pump. The Chip Cube can be operated in two modes, enrichment and analysis modes. A Large Capacity Chip II (Agilent Technologies, Santa Clara, CA) contains an enrichment 180 nL column and a 0.075x150 mm analysis column, both made of stable-bond C18 silica with 300 Å pore size packed with 5 um diameter particles. In the enrichment mode, the sample was loaded on to the enrichment column with the isocratic flow (3% B pumped by the capillary-flow pump, and then, while in analysis mode, the sample was then separated on the analytical column with the gradient elution flow pumped by the nano-flow pump. The gradient for nano-flow pump is shown in Table 1, along with other parameters for HPLC-Chip-ITMS. A 2 uL volume of digested standard BSA as quality control and the treated DPC sample solution were injected into the system separately.
MASCOT Protein Data Base Search. The MS and MS/MS spectra obtained from HPLC-Chip-ESI-ITMS were extracted and deconvoluted before they were submitted for MASCOT protein database search (Matrix Science-London, UK). They were searched under the database NCBInr. “Carbamidomethyl (C)” was selected as fixed modification, oxidation of methionine as variable modification. The MASCOT score of significance threshold was 40. The MS/MS tolerance was set as ±0.6 Da, the MS peptide tolerance as ± 1.2 Da, and peptide charges as 1+ to 3+. All the hits reported in this study have ion scores above the significance ion score thresholds.
Results and Discussion
A model DPC: the dG- Ac Lys OMe adduct. It was previously shown that under singlet oxygen oxidative conditions, 2-deoxyguanosine is oxidized forming dGox and subsequently reacts with a nucleophile (i.e. water) to form the hyper-oxidation products such as Sp and Gh. In this work, we investigate adduct formation between the amino acid N-acetyl-Lysine-Methyl ester (AcLysOMe), containing a nucleophilic side chain with dGox to produce the dG-AcLysOMe adduct. The reaction mixture was injected and separated via HPLC. To identify the retention times of the of the reactants and products, solutions containing the individual reactants, 2-deoxguanosine and AcLysOMe and the cross-linking reaction mixture were injected separately. Reactions were allowed to proceed for 1 hour, 2 hours or 4 hours before HPLC analysis. As shown in Fig. 1, the retention times of un-reacted AcLysOME and dG was determined to be 3.5 and 7 minutes, respectively. Overlaying the chromatograms from the three time points, 1, 2 and 4 hours, a peak unique to the cross-linking reaction at 5.2 minutes increased in intensity with irradiation time. This peak possesses a retention time that is halfway between that of unreacted AcLysOME and dG suggesting a combined hydrophobicity of the two components and is likely the adduct between AcLysOME and dG. This adduct was purified via fraction collection, for further analyses.
Thermolysis of the dG- Ac Lys OMe . The dG-AcLysOMe cross-link peak was collected and analyzed by flow Injection ESI-ITMS via syringe pump injections. Mass spectral analysis reveals a species with a m/z = 484.1, which is consistent with the of [Sp-5- Ac Lys OMe H]+. Fig. 2a shows the resulting mass spectra and the proposed structure of the adduct. MS2 fragmentation generates a m/z value of 386.1, which corresponds to the loss of ribose and a proton from the molecule, suggesting a nucleoside component of the adduct. The mass spectra and the proposed structure is shown in Fig. 2b.
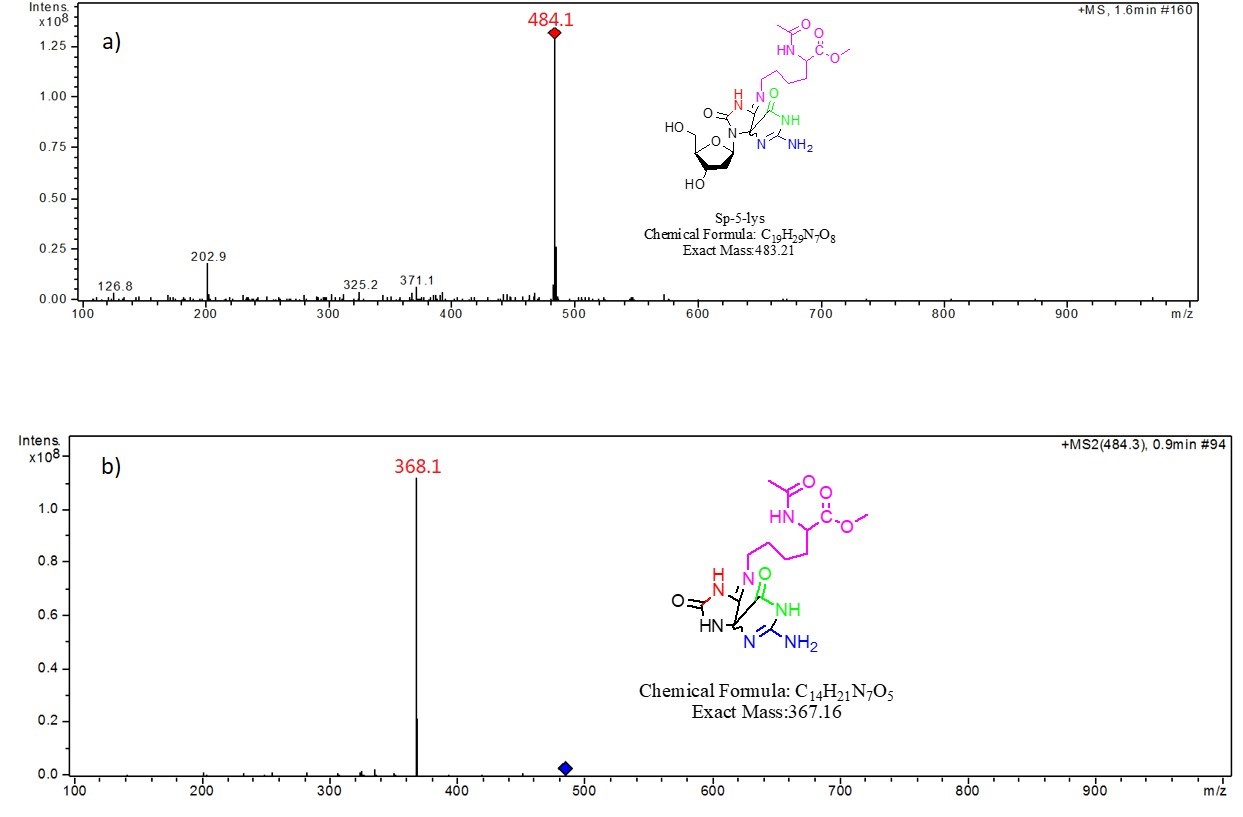
Fig. 2 a) MS of the dG-AcLysOMe cross-link compound, b) MS2 of the dG-AcLysOMe cross-link compound, both at 15 eV CID energy.
Mass spectral analysis supports the formation of Sp- AcLysOMe as the predominant product during singlet oxygen-induced oxidation, which is consistent with previous results from our group. In order to investigate the thermal stability of the cross-link covalent bond, the purified adduct was dried and heated at 170 oC for one hour, re-suspended in 50 µL doubly distilled water and injected into the ITMS system. As shown in Fig. 3, in the resulting mass spectra, m/z 484 corresponds to the intact adduct and m/z 368.1 corresponds to the adduct minus ribose. A m/z of 203, assigned as AcLysOMe, indicates a fragmentation at the covalent bond between AcLysOMe and dSp. The mechanism of cross-link formation and thermal degradation is described in Fig. 4.
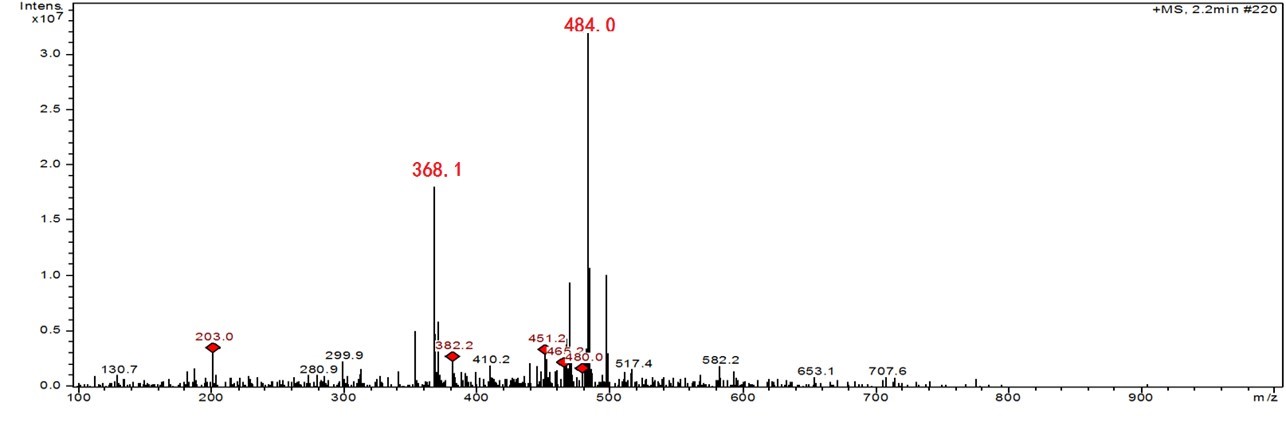
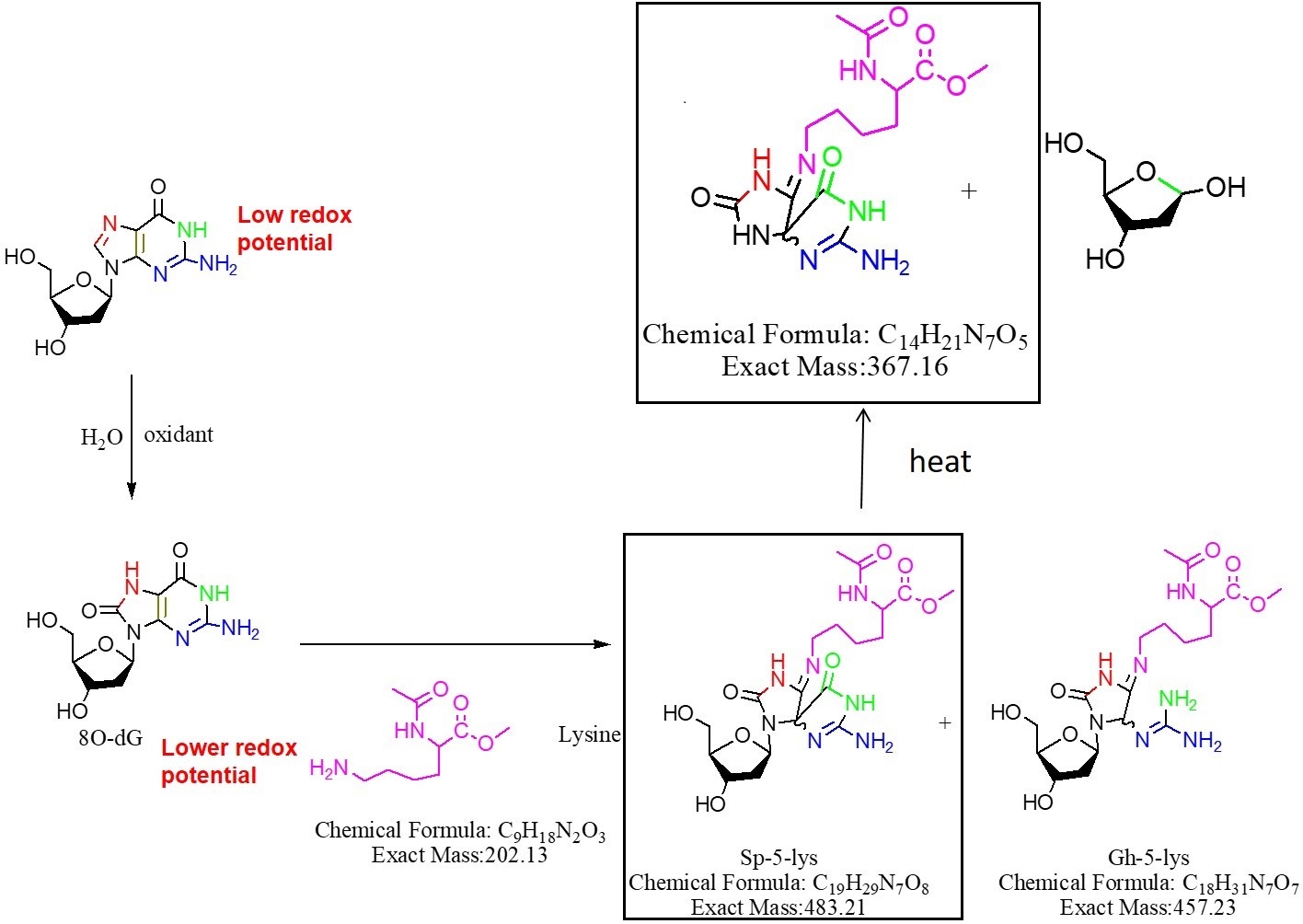
Fig. 4 Proposed reaction route for the thermal degradation of the dG-AcLysOMe adduct, the structures observed by MS analysis shown in Figure 3 are highlighted.
Identification of whole protein DPCs. Having shown that the covalent bond between AcLysOMe and dGox is thermally stable under our experimental conditions, the purified DNA-RNase cross-linking adduct was heated and hydrolyzed under the same conditions to determine if similar stability would be observed. The solution after thermal treatment was separated by SEC-HPLC with UV-Vis detection. In Fig. 5, an iso-absorbance plot of the chromatogram obtained is shown. Due to the effect of its high hydrodynamic radius (highest MW), the earliest eluting peak at 260 nm was assigned to the intact DNA-RNAse A adduct. Additionally, two other peaks are observed with the absorbance maximum around 260 nm. By comparing the elution volume with the SEC standards, the peak with a retention time of 24.2 min corresponds to the DNA strand while the peaks eluting after 30 minutes are low molecular mass de-composition products of the thermal degradation. Most importantly, the fraction eluting at 26 min showed two UV absorbance bands; one at 280 nm characteristic for protein and the second at 240 nm, matching the cross-linking bond specific absorbance. Under our experimental conditions, we conclude that this fraction exhibited the unique absorbance for protein and oxidized dG is the RNAse A-dG residue produced after thermal degradation, therefore it was of interest and was isolated via fraction collection for further studies.
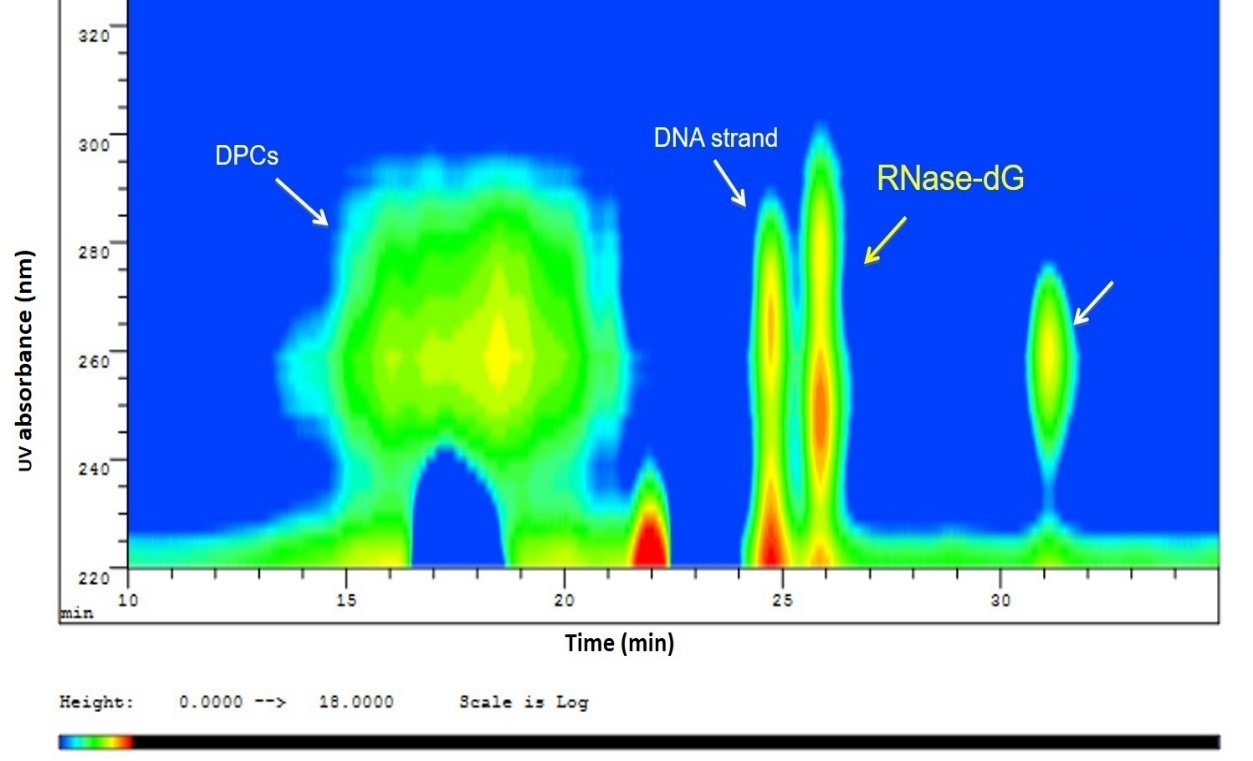
Fig. 5 Iso-absorbance plot of the SEC-UV-Vis separation after thermal degradation of DPC showing partial cleavage of the glycosidic bond, leaving a DNA strand and the Rnase-dG adduct. At the same time, some of the DPC remained stable, which eluted earlier
Identification of the dG-containing peptide-by-peptide sequence matching. The isolated dG-RNAse A adduct from the preceding section was subjected to trypsin digestion and analyzed using HPLC-Chip-ESI-ITMS. The resulting mass spectrum was used to search the MASCOT database for peptide identification. The search results are shown in Table S1. For the trypsin proteolysis of the RNase A, the maximum missed cleavage was set as 2 and the minimum peptide length was set as 5. Variable carbamidomethyl modification of Cys was accepted. The detection range was set from m/z 250 to 3000. Tryptic digest of RNAse A will result to 45 peptide fragments as predicted by the PeptideCutter search tool (http://web.expasy.org/peptide_cutter/). Lysine containing peptides are predicted to form an adduct with guanine in DNA. Addition of one dG molecule will result in Δm/z of 281.1. Because there are multiple lysine residues in the RNAse A sequence, each of these residues can potentially form adducts with dG resulting to peptide fragment cross-linking with multiple equivalents of dG. For example, if the molecular weight of one peptide is M, it is possible to observe [M-H-ndG]+, [M-H-ndG]++, and [M-H-ndG]+++. All the possibilities for the m/z value were calculated and shown in Table S1. The method of calculation is detailed in the Electronic Supplement. The extracted ion chromatogram (EIC) for each of these possibilities was checked and both MS and MS2 were also extracted to determine if any distinct peptide feature is observed.
Two values from Table S1, 810.36 m/z and 640.78 m/z fit the requirements for a cross-linked peptide. Their EIC showed an apparent peak and MS and MS2 indicated distinctive characteristics and peptide. The 810.36 m/z matched the m/z value of [M-3H-1dG] +++ , where M corresponds to the peptide, ETGSSKYPNCAYKTTQANK. Since there are three lysine residues in this peptide, dG can be cross-linked to any or all of these residues. Additionally, dG can form adducts with tyrosine residues in the sequence giving an m/z that is equal to that of dSp-Lysine adduct. To account for all the possible masses, a theoretical fragmentation table for each cross-linking possibility was constructed. The thirty-one most intense peaks during MS2 are compared with the predicted values in the fragmentation table, with the highest coverage of ten of the thirty-one peaks matching the theoretical fragmentation table for the adduct, ETGSSK(dG)YPNCAYKTTQANK. Two cross-links had low coverage, that is, 7 in one of the cross-links and 2 in the other of the 31 most intensive peaks, show a match on the fragmentation table for the MS2 from the corresponding EICs. Although 640.78 m/z fit the MS characteristics of KETAAK, further investigation shows that 0 of the 31 peaks in the MS2 spectrum matched with the theoretical fragmentation table, meaning this fit is coincidental. The MS/MS spectra and its features are shown in Fig. 6.
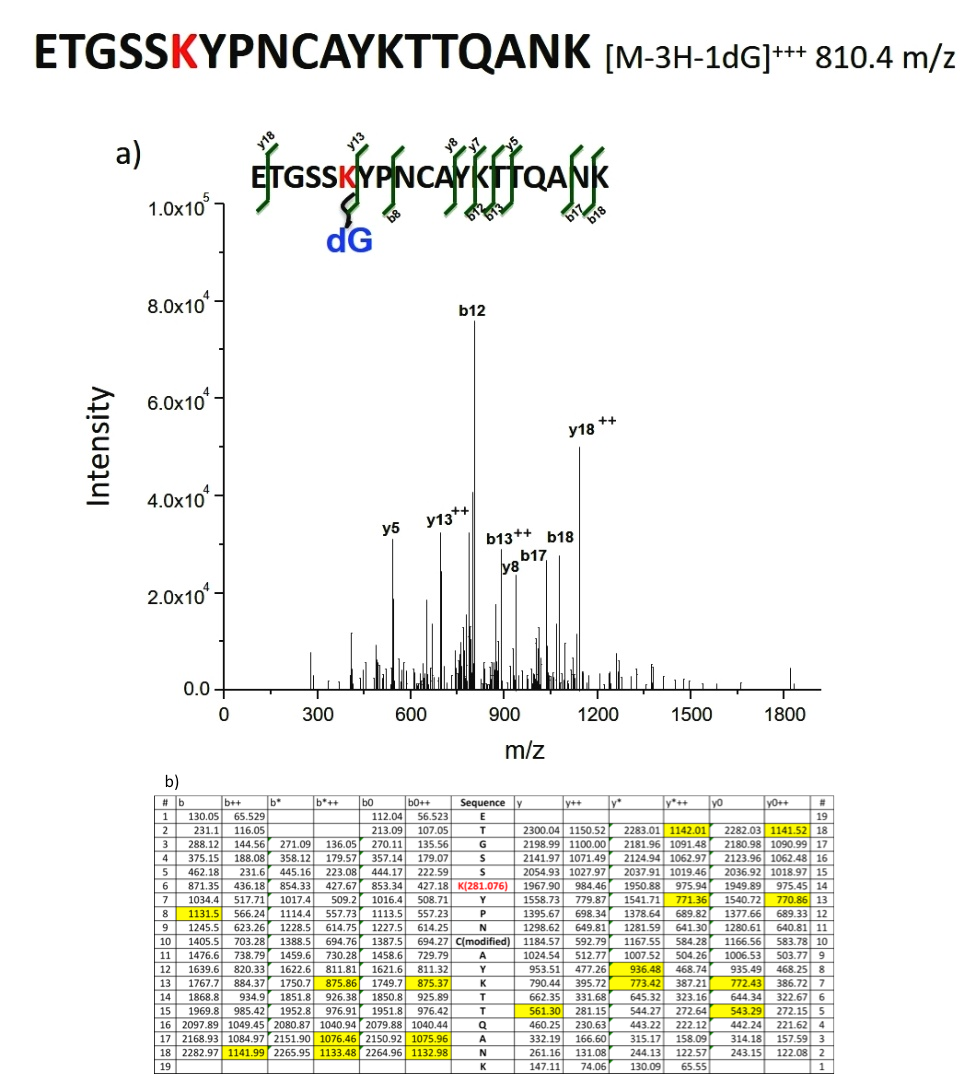
Fig. 6 a) Mass spectrum for Identification of the dG-containing peptide by peptide sequence matching. b) De novo analysis showing the calculated theoretical MS2 based on b and y ions. Highlighted are the matches to the experimental MS/MS spectrum of Fig. 6a)
Cross-linking prediction. Therefore, it can be concluded that the cross-linking reaction first took place on 91K in the peptide, ETGSSKYPNCAYKTTQANK, on the protein ribonuclease A. The structure of ribonuclease A is shown in Fig. 7 (generated with Visual Molecular Dynamics, University of Illinois, USA) and the peptide sequence is labeled in red chain with 91K shown in balls. From this structure scheme it can be seen that this lysine is exposed, making it sterically possible to associate with the DNA. Secondly, this peptide appears as a loop, which is preferable to trap the DNA.
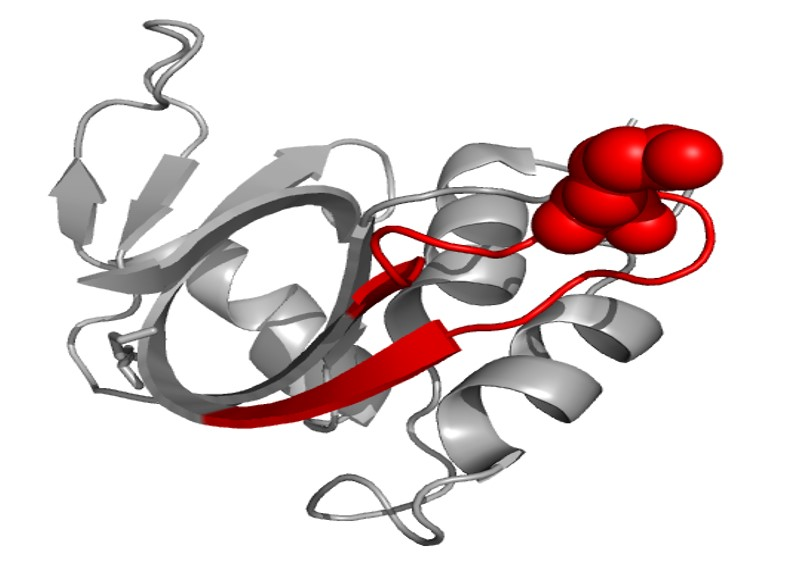
Conclusions
In this work, we demonstrate thermal stability of DNA-protein adducts, both in the nucleoside-amino acid model, and more importantly, in the DNA-protein model. These findings suggest that these lesions are stable and are not easily reversed. There are two important implications. The first one is biological in that, DNA-protein cross-link formation will have significant consequences when generated inside the cell if it remains unrepaired. It is likely to block important cellular processes such as replication (producing the highly detrimental double strand breaks that can lead to genetic instability and cancer) and can also hamper the transcription process and impede production of important proteins. The second implication of these findings is that because of the stability of the adduct, a facile method of DPC characterization (i.e. the cross-linking site, the amino acid and base involved and more importantly identifying the proteins that participate in cross-linking) can be developed. In this study, we have done the first step in intact cross-link characterization by successfully hydrolyzing the adduct between DNA and protein, removing only the un-cross-linked nucleosides. This was followed by tryptic digestion, which was initially thought to induce cross-link hydrolysis and would hinder characterization of intact DPCs by mass spectrometry. We are able to identify the cross-linking site following proteolysis of cross-linked RNAse A. This is a significant advance from the small molecule models and will be useful for total characterization of DNA-protein cross-links, protein identification and in biomarker discovery, where the cross-link identity has potential use as fingerprint for disease.

