










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.59 no.4 Ciudad de México oct./dic. 2015
Article
Electrochemical Behavior of Ni(II) Complexes with N2S2 and N6 Ligands as Potential Catalysts in Hydrogen Evolution Reaction
Vanessa Ramírez-Delgado,1 Guadalupe Osorio-Monreal,1 Luis Felipe Hernández-Ayala,2 Yolanda Reyes-Vidal,1,3 Juan Carlos García-Ramos,2‡ Lena Ruiz-Azuara2 and Luis Ortiz-Frade1*
1 Centro de Investigación y Desarrollo Tecnológico en Electroquímica S.C. Parque Tecnológico Querétaro, Sanfandila, Pedro de Escobedo, C.P. 76703. Querétaro, México. lortiz@cideteq.mx
2 Laboratorio de Química Inorgánica Medicinal. Departamento de Química Inorgánica y Nuclear, Facultad de Química, Universidad Nacional Autónoma de México, Av. Universidad 3000, Ciudad Universitaria, México, D.F., 04510, México. ‡ Currently a postdoctoral fellow at Departamento de Fisicoquímica, Instituto de Química, UNAM.
3 Catedra Conacyt -CIDETEQ.
Received October 7th, 2015;
Accepted February 17th, 2016.
Abstract
In this work, two Ni(II) complexes with the tetradentate ligand N2S2 (pdto=1,8-bis(2-pyridyl)-3,6-dithioctane,) and the hexa-dentate ligand N6 (bdahp= 2,9-bis-(2',5'-diazahexanyl)-1,10-phenan-throline) were prepared in order to explore its electrochemical behavior, that indicate their potential use as molecular catalysts for the hydrogen evolution reaction. The Ni(II)-pdto complex presented two consecutive one electron transfer [Ni(II)-(pdto)] + 1e- [Ni(I)-(pdto)] and [Ni(I)-(pdto)] + 1 e- - Ni(0) + pdto. On the other hand the Ni(II)-bdahp complex presented the electrochemical reduction Ni(II)-(bdahp) + 1e- Ni(I)-(bdahp) followed by a coupled chemical reaction in an ECi mechanism, where a de-coordination of the diiminic moiety of the bdahp ligand was proposed. It was demonstrated that the pdto ligand promotes reduction over Ni(II) at less negative reduction potential in comparison when the ligand bdahp is presented.
Key words: Electrochemistry, Ni(II) complexes; N2S2 ligand; N6 ligand.
Resumen
En este trabajo se prepararon dos complejos de Ni(II) con un ligante tetradentado N2S2 (pdto=1,8-bis(2-piridil)-3,6-ditioctano) y un ligante hexadentado N6 (bdahp = 2,9-bis-(2',5'-diazahexani-l)-1,10-fenantrolina) con el fin de estudiar su comportamiento electroquímico que indique su potencial uso como catalizadores moleculares en la reacción de evolución de hidrógeno. El complejo Ni(II)-pdto presenta dos transferencias mono-electrónicas consecutivas [Ni(II)-(pdto)] + 1e- [Ni(I)-(pdto)] y [Ni(I)-(pdto)] + 1 e- - Ni(0) + pdto. Por otro lado el complejo Ni(II)-bdahp presentó la reducción electroquímica Ni(II)-(bdahp) + 1e- Ni(I)-(bdahp) seguida de una reacción química acoplada en un mecanismo de ECi, donde se propone una des-cordinación de la parte diimínica del ligante bdahp. Se demostró que el ligante pdto promueve la reducción del Ni(II) en potencial de reducción menos negativo en comparación cuando está presente el ligante bdahp.
Palabras clave: Electroquímica; complejos Ni(II); ligante N2S2; ligante N6.
Introduction
The economic dependence of fossil combustibles has motivated scientists around the world to develop alternative energy sources. The use of solar radiation for water splitting to produce H2 and O2, also called artificial photosynthesis, and the electrochemical hydrogen evolution are considered potential strategies for energy storage [1]. Inert electrodes with noble metals, particularly bulk and nanostructured Pt, have been widely used to achieve this goal. Nevertheless coordination compounds present advantages, such as the use of non-expensive metals and the accurate control of chemical reactivity. Typically Co(II)-diglyoxime complexes in non-aqueous solvents with HA proton donors have been used for hydrogen evolution catalysis [2-6]. The low stability of these systems has motivated to explore alternative compounds such as Co(II)-polypyridine complexes and highly competitive Ni(II) complexes [6-10]. DuBois designed the Ni(II) complex [Ni(PPh2NPh)2](BF4)2 (PPh2NPh = 1,3,6-triphelyl-1-aza-3,6-diphopahcyclo heptane) with a ligand containing phosphorus donor atoms and a pendant protonable amine as proton relays for the electrochemical reduction of [(DMF)H] OTf, pKa=6.1 in MeCN [11]. Other examples of Ni(II) complexes, [Ni(7PPh2NC6H4X)2](BF4)2 (7PPh2NC6H4X = 1-para-X-phenyl-3,6-triphenyl-1-aza-3,6-diphos- phacycloheptane, X= OMe, Me, Br, Cl or CF3, with similarities to the ligand PPh2NPh were also reported [12-16]. The basicity of the amine in the ligand, and the catalytic activity was modified by the presence of electron donating and electron withdrawing substituents X on the aromatic ring attached to the nitrogen atom. In a biomimetic approach to the Ni-Fe Hydrogenases, ligands with N/S donor atoms, specifically substituted and non-substituted pyri-dine-2-thiolate and pyrimidine-2-thiolate were also used to prepare Ni(II) complexes, with a decent electro-catalysis and photo-driven hydrogen evolution activities [15-20]. From all these studies it was figured out that in the proposed mechanism, the first step is a one electron uptake over Ni(II), followed by the formation of hydride species Ni(II)-H. From this work, it was established that for a good catalyst, flexible ligands are desirable because of the preferential geometries of the proposed Nickel oxidation states in the catalytic cycle. Another important factor is the reduction potential of the couple Ni(II)/Ni(I). A more negative value increases the hydride donor abilities of the Nickel-hydrides, making the process more efficient [12].
A good candidate for a potential hydrogen evolution catalyst is the Ni(II) complex with the ligand 1,8-bis(2-pyr-idyl)-3,6-dithiaoctane (pdto), due to the presence of N/S donor atoms and the already demonstrated high flexibility toward the preferential geometry of a metal center, see scheme 1 [21-40]. On the other hand, the negative reduction potential of Ni(II) coordinated to a diiminic moiety and the flexibility of proton-able open chain amines, make that the Ni(II) complex with the ligand 2,9-bis-(2',5'-diazahexanyl)-1,10-phenanthroline (bdahp), see scheme 1, can be considered as a possible catalyst for hydrogen evolution reaction. Therefore, in this work we prepared two Ni (II) complexes with flexible ligands N2S2 (pdto) and (bdahp) (pdto=1,8-bis(2-pyridyl)-3,6-dithioctane, bdahp= 2,9-bis-(2',5'-diazahexanyl)-1,10-phenanthroline) in order to explore its electrochemical behavior that provides information necessary to propose their potential use as catalysts for hydrogen evolution reaction.
Results and discussion
Characterization of [Ni(pdto)(CH3CN)2](BF4)2
The reaction between the Nickel salt [Ni(H2O)6](BF4)2 and the ligand 1,8-bis(2-pyridyl)-3,6-dithioctane yield a blue powder with empirical formula C20H26N4S2B2F8Ni. The IR spectrum displays typical adsorption bands for pdto. The stretching vibrations from pyridine rings v(C=C) + v(C=N) are recorded at 1608 and 1565 cm-1. The asymmetric stretching vas(CH) and symmetric stretching vs (CH) are observed at 2855, 2835 and 2819 cm-1. Methylene scissoring bands (δs CH2) occur at 1436 and 1482 cm-1. Sharp absorption bands at 3103 and 3016 cm-1 attributable to aromatic stretching v (=C-H) are also observed. The absorptions bands corresponding to the out of plane δ(=C-H) bending is registered at 770 cm-1. Characteristic signals for coordinated CH3CN, v(-C-H) and v(C=N are recorded at 2950, 2920, 2257 and 2287 cm-1. A broad signal around 1080 cm-1 characteristic for BF4- anion was observed. With this information it is not possible to propose a geometry for the Ni(II) complex. Thus NIR diffuse reflectance spectra was acquired, see fig. 1. Three absorption bands at 10535, 16813 and 26650 cm-1 can be observed, which correspond to the electronic transitions v1= 3A2g (F) → 3T2g (F); v2 = 3A2g (F) →3T2g(F); v3 = 3A2g (F) -3T1g (P) for typical octahedral Ni (II) complexes. These are in agreement with calculated effective magnetic moment (|ieff) 2.97 MB (2 unpaired electrons) [41-43]. Additionally a Charge Transfer (CT) absorption band at 30337 and a spin-forbidden electronic transition 3A2g→ 1Eg at 9655 cm-1 were also recorded [44]. A 10 Dq value of 10535cm1 is assigned, according to the literature [45]. The above discussion allows us to propose unequivocally the molecular formula [Ni(pdto)(CH3CN)2](BF4)2.
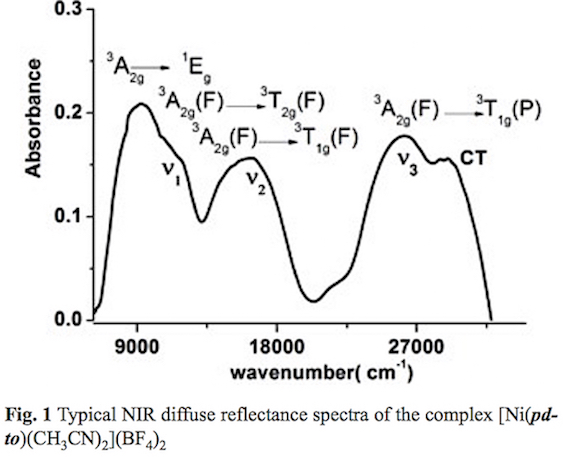
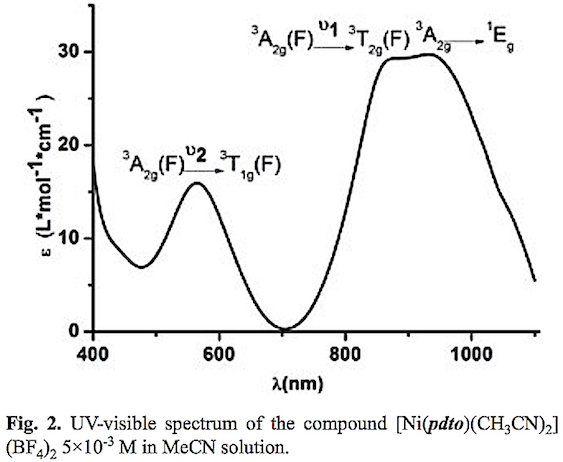
On the other hand, to explore the geometry of this Ni(II) complex in solution, conductimetric and spectroscopic measurements were performed. A 1 mM solution of the complex in acetonitrile presented a molar conductance (Λm) value of 320 Ω2-1cm2mol-1, characteristic for a 2:1 electrolyte. The UV-visible spectrum of the compound in acetonitrile shows the electronic transitions v2=3A2g(F) → 3T1g (F) , v1= 3A2g(F) → 3T2g(F), and 3A2g→ 1Eg at 565 nm (17699 cm-1), 880 nm (11360 cm-1) and 936 nm (1070 cm-1) respectively, see fig. 2. These results indicate that the complex present an octahedral geometry with the same donor atoms around the Ni(II) center in acetonitrile solution and in solid state. The slight changes in maximum absorption bands are related to solvation effects.
Characterization of [Ni(bdahp)](PF6)2
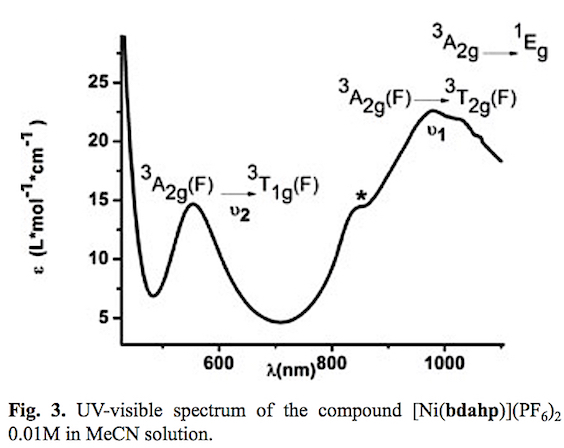
A purple powder was obtained with empirical formula NiC20H28N6P2F12. The presence of the polypyridine 1,10-phen-athroline moiety and aliphatic amines of the bdahp ligand are confirmed by IR spectroscopy. The stretching absorption bands v (C=C) + v (C=N) occur at 1591, 1500, 1462 and 1435 cm-1. The aromatic stretching v (=C-H) signals are detected at 3087 cm-1. The absorptions bands corresponding to the out of plane C-H bending are registered at 743, 723, and 686 cm-1. The (N-H) stretching bands are observed at 3344 and 3323 cm-1. The absorption bands for the stretching v (=C-H) associated to CH3 and CH2 groups are detected at 2997, 2970, 2945 and 2925 cm-1. Moreover the counter ion (PF6-) was recorded around 825 cm-1. The electronic spectra of this complex in acetonitrile solution is presented in fig. 3. In the same way that in the Ni(II)-pd-to complex the effective magnetic moment (μeff) 3.04 BM and the electronic transitions in acetonitrile solution v2=3A2g(F) → 3T1g(F), v1= 3A2g (F) → 3T2g (F) and 3A2g → 1Eg at 551 nm (18148 cm-1) , 980 nm (10204 cm-1) and 1017 nm (9832 cm-1) demonstrate a octahedral geometry for the Ni(II) complex. The presence of a shoulder (*) around 830 nm, is attributed to electronic transitions for RHN-Ni(II) fragment which has a lower crystal filed splitting according to spectrochemical series [42]. The solution behavior was complemented with conductimetric measurements. The calculated molar conductance value Λm= 300 Ω-1cm2mol-1 for a 1 mM solution of the complex (2:1 electrolyte) indicates the complete dissociation of the compound that generate the cationic complex and two counterions PF6-. This information allows the proposal of molecular formula [Ni(bdahp)](PF6)2.
Electrochemical behavior of [Ni(pdto)(CH3CN)2](BF4)2 in acetonitrile solution
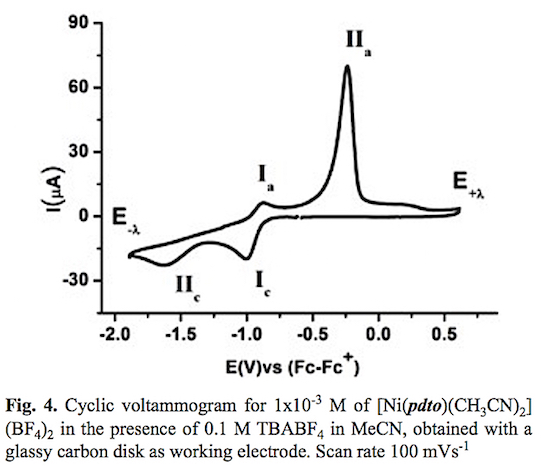
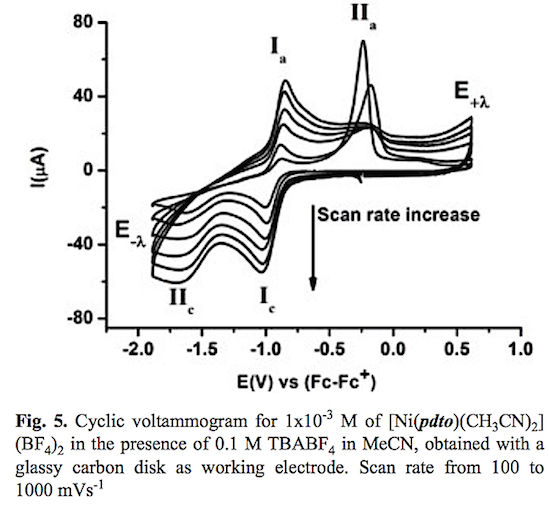
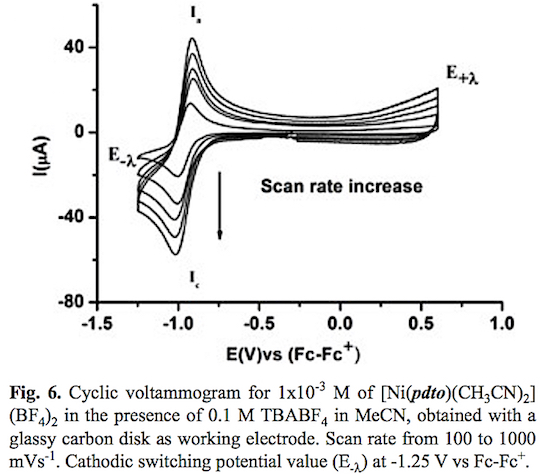
A typical cyclic voltammogram of the complex [Ni(pdto) (CH3CN)2](BF4)2 in acetonitrile solution in the presence of 0.1 M TBABF4 is shown in fig. 4. When the potential scan was started from open circuit potential to negative direction two reduction processes Ic and IIc and two oxidation processes Ia and IIa were recorded. The corresponding potential peaks for these signals are Epc(I) = -1.004 V, Epc(II) -1.630, Epa(I) = -0.889 V and Epa(II) = -0.237 V vs Fc-Fc+. When the scan rate was increased, see fig. 5, the signals Ic, IIc and Ia presented an increment on their corresponding current values. The opposite was registered for signal IIa. It should be highlighted that an increase of current for signals Ia and Ic when the time scale of the experiment was diminished, indicates a dependence between them. To prove this idea cyclic voltammetry experiments using a lower cathodic switching potential value (E-λ) were carried out. Fig. 6 shows a series of voltammograms at variable scan rate with a E-λvalue at -1.25 V vs Fc-Fc+, where only the redox processes Ic and Ia were observed. The anodic peak potential Epa(I) present a shift from -0.889 (long E-λ ) to -0.924 (short E-λ) vs Fc-Fc+, meanwhile no change was observed in the cathod-ic peak potential Epc (I). The ratio ipa(Ia)/ipc(Ic) presents a value close to the unity in the whole range of scan rates. Therefore we propose the electron transfer Ni(II)-(pdto) + 1 e- Ni(I)-(pdto) for process I, with a half wave potential (E1/2) value of -0.964 V vs Fc-Fc+ and a difference between potential peaks (ΔEp) close to 0.080 V. Considering the assignation for process I and cyclic voltamograms, it was possible to attribute signal IIc to the electrochemical reaction Ni(I)-(pdto) + 1 e- - Ni(0) + pdto, and process IIa to the anodic dissolution of Ni(0) deposited in the forward scan. This fact explain the change in the anodic peak potential values Epa for process Ia when the switching was modified, in terms of different energetic requirements for the oxidation of the Ni(I)-(pdto) species over glassy carbon (short E-λ) and over a Ni(0) deposit (long E-λ). One step chronoamper-ometry experiment stepped from open circuit potential (Ei=0) to potential value E1= -1.247 V vs Fc-Fc+ with a perturbation time τ =1 s, allows to calculate a diffusion coefficient of 1.443 *10-5 cm2 s-1 according to Cottrell's law [46, 47].
Electrochemical behavior of [Ni(bdahp)3](PF6)2 in acetonitrile solution
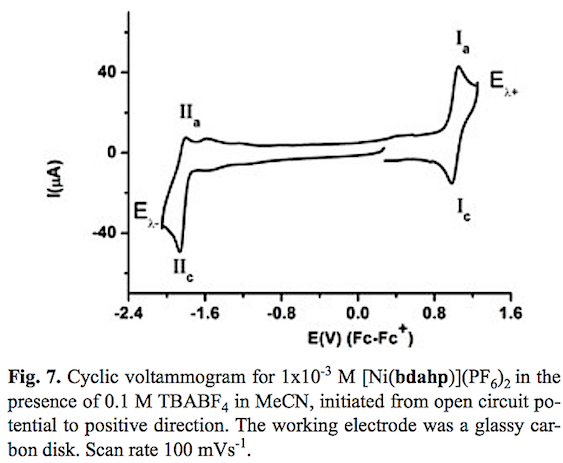
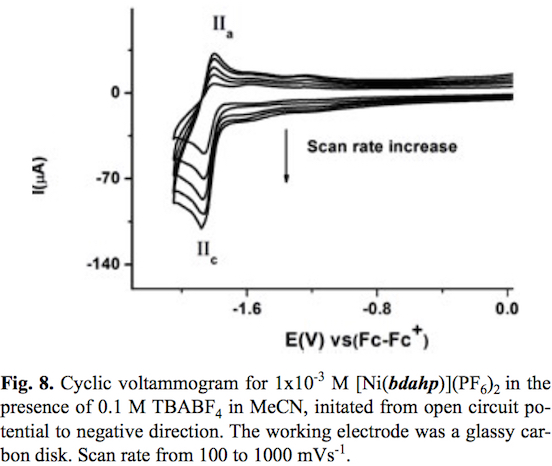
The cyclic voltammogram of compound [Ni (bdahp)3](PF6)2 obtained with a glassy carbon electrode at scan rate v=100 mV s-1 in acetonitrile solution is shown in fig. 7. In the complete scan (started from open circuit potential to negative direction) the voltammogram displays two oxidation signal Ia and IIa with their corresponding reduction signal Ic and IIc. For process I a half-wave potential (E1/2) value of 1.013 V/Fc-Fc+ and a ΔEp value of 0.060 V value was obtained. On the other hand for process II their corresponding ΔEp and E1/2 values were 0.060 V and -1.832 V/Fc-Fc+ respectively. In order explain the nature of both electron transfers, the electrochemical study of the complex [Ni(1,10-phen)3](BF4)2 in the same experimental conditions was carried out. One oxidation process with a half wave potential E1/2 = 1.419 V vs Fc-Fc+ associated to [NiII(1,10-phen)3]2+ [NiIII(1,10-phen)3]3++1e- was observed [48,49]. A second process associated to the reduction [NiII (1,10-phen)3]2+ +1e [NiI(1,10-phen)3]+ with a half wave potential E1/2 = -1.629 V vs Fc-Fc+ was also detected. Taking into account similar oxidizable and reducible groups between [Ni(bdahp)]2+ and it was possible to establish the electrochemical reactions; [Nin(bdahp)]2+[Nim(bdahp)]3++1e- for process I and [NiII(bdahp)]2++1e [NiI(bdahp)]+ for process II. This is in agreement with the fact that imine ligands such as 1,10-phen and bipyridine offer a more attractive environment for Ni(I) and other metal complexes in low oxidation state by allowing electron delocalization over the ligand n system [49-55]. When the scan rate was increased the corresponding cyclic voltammograms showed a better definition of the oxidation signal IIa with an increment in its current value. The ratio ipa(IIa)/ipc(IIc) presented small values at low scan rate, characteristic of a coupled chemical reaction in an ECi mechanism, see fig. 8. In literature it has been stated that the electrochemical reduction for the cationic complexes [NiIIL3]2+ (L=1,10-phen, and bipyridine) in anhydrous acetonitrile results in [NiIL2]2+ [49,56,57]. Based on this fact we can propose a de-coordination of the diiminic moiety of the bdahp ligand with the electrogenerated Ni(I) forming the species [NiI(κ-4N-bdahp)]+, detectable in the time scale of the cyclic voltammetry employed. According to the literature the rate constant (kf) for the ECi mechanism was calculated [58,59]. The obtained values were 0.160 ±0.007 and 0.170 ±0.001 for compounds [Ni(1,10-phen)3]2+ and [Ni(bdahp)]2+ respectively. The low value for the 1,10-phen complex in comparison with the bdahp complex is related to high chelate effect.
From one step chronoamperometry experiments, with a potential step from open circuit potential to -1.90 vs Fc-Fc+, a diffusion coefficient of 7.97x10-5 cm2s-1 was calculated according to Cottrell law. Similar experiments were performed for the complex [Ni(1,10-phen)3]2+ stepping the potential to -1.80 vs Fc-Fc+ (Do=3.48874>10-5 cm2s-1).
Comparison of the electrochemical behavior of Ni(II) complexes
Fig. 9 presents a graphical comparison of the electrochemical reductions of the compounds studied in this work. From this figure it can be established that the Ni(II) complex with the ligand N6 [Ni(bdahp)]2+ which contains a diiminic moiety and flexible aliphatic amines the redox potential is shifted to more negative values in comparison with the [Ni(pdto)(CH3CN)2]2+ complex. The change in denticity of a bidentate ligand (1,10-phenanthroline) to a hexadentate ligand N6 bdahp= 2,9-bis-(2',5'-diazahexanyl)-1,10-phenanthroline causes that the reduction of Ni(II) requires more energy, despite the fact that the tris-chelate [Ni(1,10-phen)3] 2+ complex should present a more negative reduction potential, according to the additivity contribution of three ligands with high n acceptor character [60]. Hence this shift in reduction potential could be attributable to the chelate effect. An important change in the diffusion coefficient could be observed, as a consequence of the molecular size of the cationic complexes considering the solvation sphere of each compound. Table 1 shows a summary of the electrochemical reduction processes of the compounds studied in this work, and other Ni(II) complexes used for electrochemical hydrogen evolution taken from literature.
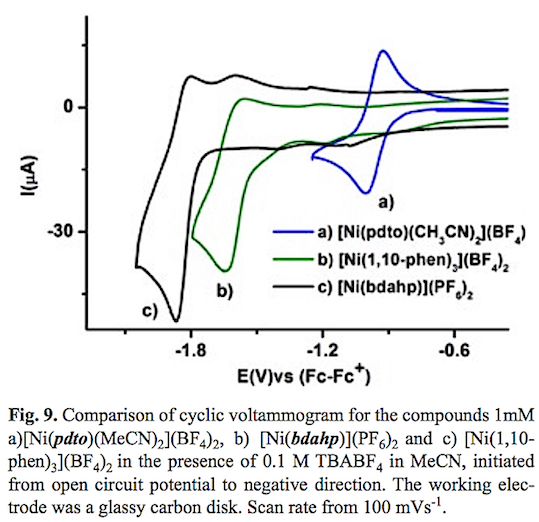
The complex [Ni(bdahp)]2+ presented a more negative reduction potential (-1.83 vs Fc-Fc+) than those reported for Ni(II) complexes with ligands containing phosphorus donor atoms on its structure [Ni(PPh2NPh)2]2+ (PPh2NPh = 1,3,6-triphelyl-1-aza-3,6-diphopahcyclo heptane) and [Ni(7PPh2NC6H4X)2]2+ (7PPh2NC6H4X =1-para-X-phenyl-3,6-triphenyl-1-aza-3,6-diphosphacycloheptane, X= OMe, Me, Br, Cl or CF3) (from -1.13 to 1.05 vs Fc-Fc+). Considering that a negative redox potential value increases the hydride donor ability, making the hydrogen evolution reaction more efficient, a good catalytic activity for the complex [Ni(bdahp)]2+ is expected. On the other hand the reduction potential (-0.96 vs Fc-Fc+) of the cationic complex [Ni(pdto)(MeCN)2]2+ suggests a low hydride donor ability in comparison with the reported values for the bioinspired Ni(II) complexes with N/S ligands [Ni(X-pyS)3]-, [Ni(pySH)4]2+, [Ni(4,6-Y2-pymS)3]- and [Ni(4,4'-Z-2,2'-bpy)(pyS)2] (pyS = pyridine-2-thiolate, X= 5-H, 5-Cl, 5-CF3, 6-CH3), (pymS = pyrimidine-2-thiolate Y= H, CH3, Z = H, CH3, OCH3 and bpy = bipyridine) ( from -1.73 to 2.01 vs Fc-Fc+). However the Ni(II)-pdto system presented a necessary reversible electron transfer Ni(II) + 1 e- Ni(I) in a catalytic cycle, which is not observed in the Ni(II) complexes with substituted and non-substituted pyridine-2-thiolate, pyrimi-dine-2-thiolate and 4,4-bipyridines, see table 1.
Conclusions
Using typical characterization techniques octahedral Ni(II) complexes with the tetradentate ligand N2S2 (pdto=1,8-bis (2-pyridyl)-3,6-dithioctane,) and the hexadentate ligand N6 (bdahp = 2,9-bis-(2',5'-diazahexanyl)-1,10-phenanthroline) are proposed . The cationic complexes [Ni(pdto)(CH3CN)2]2+ and [Ni(bdahp)] 2+ presented an electron transfer from Ni(II) to Ni(I), with half wave redox potential controlled by donor atoms. It was demonstrated that the coordination of a N2S2 ligand with Ni(II) makes the reduction potential shift to less negative values in comparison to Ni(II) complexes with a N6 ligand bdahp. The electrochemical behavior, through an analysis of the reduction potential values of [Ni(pdto)(CH3CN)2]2+ and [Ni(bdahp)]2+ suggest that both complexes can be considered as potential catalysts for hydrogen evolution reaction in a future work.
Experimental section
Chemicals
All chemicals and solvents in this work were used as received from Aldrich Chemical Co., Acros Organics and J.T. Baker.
Synthesis of the ligand 1,8-Bis(2-pyridyl)-3,6-dithioctane (pdto)
The ligand 1,8-bis(2-pyridyl)-3,6-dithioctane (pdto) was prepared using the method described by Goodwin and Lions. [21]. Yield 70%, elemental analysis calculated for C16H20N2S2: calculated %C 63.1, %H 6.6, %N 9.2, %S 21.1; found: %C 63.1, %H 6.2, %N 9.7, S 20.5.
Synthesis of the ligand 2,9-Bis-(2',5'-diazahexanyl)-1,10-phenanthroline (bdahp)
12.71 mmol of 2,9-diformyl-1,10-phenanthroline was dissolved in 200 lM of a (1 :1) mixture CHCl3-MeOH. Separately 38.15 mmol of N-methylethylenediamine was added to 50 mL of the CHCl3-MeOH mixture, to be added to the first solution. The reaction mixture was heated and stirred under reflux for 2 hours. When the reaction mixture was cooled at room temperature, 50.84 mmol of the reducing agent NaBH4 was added. The new reaction mixture was stirred again for 2 hours and a green solution was obtained. Extractions with CHCl3 were done and the organic phase was collected to be concentrated and re-dissolved in 5 mL of concentrated hydrochloric acid with the addition of EtOH. A pale yellow product was obtained filtered and dried. Anal. Calc. for C20H32N6Cl4 (M.W.= 498.32 g mol-1) %C, 47.79; %H, 6.52; %N, 17.23. Found: %C, 48.0, %H 6.47; %N 16.86. 1H-NMR (300 MHz, D2O) δ 7.53 (d, J= 8.35, 1H), 6.88 (d, J=8.36 Hz, 1H), 6.84 (s, 1H), 3.83 (s, 2H), 2.64 (dd, J1= 8.15, J2= 6.05 Hz, 2H), 2.51 (dd, J1= 8.31, J2= 6.46 Hz, 2H), 1.7 (s, 3 H). 13C-NMR (300 MHz, D2O) δ= 151, 143, 139, 129, 127, 123, 52, 44, 43, 33.
Synthesis of the complexes
[Ni(pdto)(CH3CN)2](BF4)2. The synthesis was carried out by dissolving 2 mmol of the metallic salt Ni(BF4) •6H2O in 25 mL of acetonitrile. Then it was added dropwise the metallic salt solution 2 mmol of pdto dissolved in 5 mL of acetonitrile. A color change from green to blue was observed. The reaction mixture was heated and stirred for 2 hours. Solvent was removed by slow evaporation until a blue powder was observed. The product was filtered and washed with ethyl ether. Elemental analysis for C20H26N4S2B2F8Ni (P. M.=618.87 g mol-1); Calc.: %C 38.81, %H 4.23, %N 9.05, %S 10.36; Found: %C 38.81, %H 4.23, %N 9.05, %S 10.36. Λm (MeCN): 320 Ω-1 cm2 mol-1. μeff: 2.907 MB (2 unpaired electrons).
[Ni(1,10-phenantroline)3](BF4)2. This synthesis was carried out by mixing 1 mmol of Ni(BF4)2 •6H2O (1>10-3 M), dissolved in 20 mL of CH3OH with 3 mmol of the ligand previously dissolved in CH3OH. A color solution change from green to pale pink was observed. The reaction mixture was stirred at room temperature for 1 hour. The solvent was removed and a pale pink precipitate was obtained to be filtered and washed with ether.
[Ni(bdahp)](PF6)2. 10 mL of aqueous solution containing 1 mmol of [H4(bdahp)]Cl4 were added to 15 mL containing 1 ;mmol of NiCl2 in aqueous solution. The pH of the solution was adjusted with NaOH. The reaction mixture was stirred for 2 hours. After this time a saturated solution of TBABF4 was added. A pale purple precipitate was obtained. The product was filtered and washed with ethyl ether. Anal. Calc. for NiC20H28N6P2F12 (M.W. = 701.01 g mol-1) %C, 34.26; %H, 4.03; %N, 12.06. Found: %C, 33.97; %H, 3.99; %N, 12.06. Λ (MeOH) = 222.6 cm2 ohm-1 mol-1. μeff = 3.04 BM.
Physical measurements
Elemental analysis of the compounds was performed with a Fissons Instruments Analyzer model EA 1108, using a sulfanilamide standard for the equipment calibration. Magnetic susceptibility measurements were obtained with Magnetic Balance Johnson Mathey MSB-1. Solid state UV-vis-NIR spectra were acquired with a Cary-5E Varian spectrophotometer 40000-4000 cm-1. IR, spectra were obtained with a Thermo-Nicolet AVATAR 320 FT-IR 400-4000 cm-1, in transmittance mode on KBr disk. Solution electronic spectra was recorded with Thermo-Scientific Evolution Array spectrophotometer (200-1100 nm), using a quartz cell (l=1cm). Conductivity measurements were obtained with Corning Pinacle 524 conductimeter (cell constant= 1 cm-1). A VARIAN Unity Inova spectrometer was used to record NMR spectra, 1H (300 MHz) and 13C (75.5 MHz) and 13C (75.5 MHz). TMS was used as a reference; CD-Cl3-D3COD was used as solvent.
Electrochemical experiments
Electrochemical experiments were performed with a potentiostat/galvanostat Biologic SP-50 using 1x10-3 M of each Ni(II) compound in acetonitrile solution + 0.1M 0.1 M TBABF4. A three cell array was used; working electrode, glassy carbon (Φ= 3 mm), counter electrode a platinum wire and as pseudo-reference a silver wire. Before each experiment the solutions were bubbled with N2, and the working electrode was cleaned by polishing its surface with a Alumina (0.3 //m), to be washed with water and sonicated. The potentials are reported vs the couple Fc/Fc+ according to the IUPAC convention [61]. Cyclic voltammetry was performed from open circuit potential to negative direction with variable scan rate from 100 to 1000 mVs-1. IR compensation was applied using the current interrupt method. Single pulse cronoamperommetry experiments were obtained stepping the potential from open circuit potential to a potential value where the electrochemical process was limited by diffusion, established from CV experiments. A time width (τ) of 1s was used.
Acknowledgement
The authors thank CONACyT (130500), UNAM-PAPIIT (217613) and UNAM-PAIP (3590-19) for financial support; VRD, and JCGR thank CONACyT and RED FARMED/CONACyT for the scholarships.
References
1. Berardi, S.; Drouet, S.; Francàs, L.; Gimbert-Suriñach, C.; Guttentag, M.; Richmond, C.; Stolla, T.; Llobet, A., Chem. Soc. Rev., 2014, 43, 7501-7519. [ Links ]
2. Eckenhoff, W.T.; McNamara, W.R.; Du, P.; Eisenberg, R., Biochim. Biophys. Acta, 2013, 1827, 958-973. [ Links ]
3. Losse, S.; Vos, J.G.; Rau, S., Coord. Chem. Rev., 2010, 254, 2492-2504. [ Links ]
4. Dempsey, J. L.; Brinschwin, B.S.; Winkler, J. R.; Gray, H. B., Accounts Chem. Res., 2009, 42, 12, 1995-2004. [ Links ]
5. Du, P.; Schneider, J. ; William, G.L. ; Brennesel, W. ; Eisenberg R., J. Am. Chem. Soc., 2009, 48, 4952-4962. [ Links ]
6. Jacques, P.-A.; Artero, V.; Pécaut, J.; Fontecave, M., P. Nat. Acad. Sci. USA, 2009, 106, 49, 20627-20632. [ Links ]
7. Tong, L.; Kopecky, A.; Zong, R.; Gagnon, K.J.; Ahlquist, M.S.G.; Thummel, R.P., Inorg. Chem., 2015, 54, 7873-7884. [ Links ]
8. Sasaki, Y.; Kato, H.; Kudo, A., J. Am. Chem. Soc., 2013, 135, 5441-5449. [ Links ]
9. Queyriaux, N.; Jane, R.T.; Massin, J.; Artero, V.; Chavarot-Kerlidou, M.; Queyriaux N., Coord. Chem. Rev., 2015, 46, 1-17. [ Links ]
10. Chen, X.; Ren, H.; Peng, W.; Zhang, H.; Lu, J.; Zhuang, L., J. Phys. Chem., 2014, 118, 20791-20798. [ Links ]
11. Helm, M.L.; Stewart, M.P. ; Bullock, R.M.; DuBois, M.R.; DuBois, D.L., Science 2011, 333, 863-866. [ Links ]
12. DuBois, D. L., Inorg. Chem., 2014, 53, 3935-3960. [ Links ]
13. Stewart, M. P.; Ho, M-H.; Wiese, S.; Lindstrom, M.L.; Thogerson, C.E.; Raugei, S.; Bullock, R.M.; Helm, M.L., J. Am. Chem. Soc. 2013, 135, 6033-6046. [ Links ]
14. Gross, M.A.; Reynal, A.; Durrant, J.R.; Reisner, E., J. Am. Chem. Soc., 2014, 136, 356-366. [ Links ]
15. Thoi, VS.; Sun, Y.; Long, J.R.; Chang, C.J., Chem. Soc. Rev., 2013, 42, 23-88. [ Links ]
16. Eckenhoff, W.T.; Eisenberg, R.; Dalton Trans., 2012, 41, 13004-13021. [ Links ]
17. Han, Z.; Shen, L.; Brennessel, W.W.; Holland, P.L.; Eisenberg, R., J. Am. Chem. Soc., 2013, 135, 14659-14669. [ Links ]
18. Das, A.; Han, Z.; Brennessel, W.W.; Holland, P.L.; Eisenberg, R., ACS Catal., 2015, 5, 1397-1406. [ Links ]
19. Xu, You; Xu, R., Appl. Sur. Sci., 2015, 351, 779-793. [ Links ]
20. Wang, M.; Han K.; Zhang, S.; Sun L., Coord. Chem. Rev., 2015, 287, 1-14. [ Links ]
21. Goodwin, H.A.; Lions, F., J. Am. Chem. Soc., 1960, 82, 5013-5023. [ Links ]
22. Brubaker, G.R.; Brown, J.N.; Yoo, M.K.; Kinsey, R.A.; Kutchan, T.M. ; Mottel, E.A., Inorg. Chem., 1979, 18, 299-302. [ Links ]
23. Manzanera Estrada, M.; Flores-Alamo, M.; Grevy. M., J.-M.; Ruiz-Azuara, L.; Ortiz-Frade, L.; Acta Crystallogr. E, 2012, 68, m135. [ Links ]
24. Ortiz-Frade, L.; Manríquez, J.; González, I.; Moreno.-Esparza, R.; Ruiz-Azuara, L., Polyhedron, 2010, 29, 328-332. [ Links ]
25. Ortiz-Frade, L.A.; Ruiz-Ramírez, L.; González, I.; Marín-Becerra, A.; Alcarazo, M.; Alvarado-Rodriguez, J.G.; Moreno-Esparza, R., Inorg. Chem., 2003, 42, 1825-1834. [ Links ]
26. Popovitch, J.M.; Addison, A.W.; Butcher, R.J.; Prushan, M.J., J. Chem Crystallogr., 2012, 42, 295-298. [ Links ]
27. Castineiras, A.; Paredes, M.; Hiller, W., Acta Crystallogr. Sect. C: Cryst. Struct. Commun., 1984, 40, 2078-2079. [ Links ]
28. Castineiras, A.; Hiller, W.; Strahle, J.; Paredes, M.; Sordo, J., Acta Crystallogr. Sect. C: Cryst. Struct. Commun., 1985, 41, 41-43. [ Links ]
29. Kolotilov, S.V.; Goreshnik, E.A.; Pavlishchuk, V.V.; Yatsimirskii, K.B., Russ. J. Inorg. Chem.,2000, 45, 967-975. [ Links ]
30. Castineiras, A.; Diaz, G.; Florencio, F.; García-Blanco, S.; Martínez-Carrera, S.J., Cristallogr. Spectrosc. Res., 1988, 18, 395-401. [ Links ]
31. Castineiras, A.; Diaz, G.; Florencio, F.; García-Blanco, S.; Martínez-Carrera, S., Z. Anorg. Allg. Chem., 1988, 101-110, 567. [ Links ]
32. Humphrey, D.G.; Fallon, G.D.; Murray, K.S., J. Chem. Soc. Commun., 1988, 1356-1358. [ Links ]
33. Rajendiran, V.; Murali, M.; Suresh, E.; Sinha, S.; Somasundaram, K.; Palaniandavar, M., Dalton Trans., 2008, 1, 148-163. [ Links ]
34. Castiñeiras, A.; Molleda, C.; Masaguer, J.R.; Coto, V., Transition Met. Chem, 1984, 8, 129-131. [ Links ]
35. Ramírez-Delgado, V.; Morales León, R.E.; Hernández-Ayala, L.F.; Ramírez Coutiño, V.A.; Rodríguez, F.J.; Osorio-Monreal, G.; García-Ramos, J.C.; Flores-Alamo, M.; Ruiz-Azuara, L.; Ortiz-Frade, L., Polyhedron, 2014, 74, 72-78. [ Links ]
36. Pavlischchuk, V.V.; Koltilov, S.V.; Michael, E.S.; Prushan, J.; Addison, A.W., Inorg. Chim. Acta, 1998, 278, 217-222. [ Links ]
37. Pavlishchuk, V.V.; Kolotilov, S.V.; Addison, A.W.; Sinn, E.; Prushan, M.J., Russ. J. Inorg. Chem., 2000, 45, 544-550. [ Links ]
38. Bermejo, E.; Castineiras, A.; Dominguez, R.; Strahle, J.; Hiller, W., Acta Crystallogr. Sect. C: Cryst. Struct. Commun., 1993, 49, 324-326. [ Links ]
39. Worrell, J.H.; Genova, J.J.; Dubois, T.D. J. Inorg. Nucl. Chem., 1978, 40, 441-446. [ Links ]
40. Bermejo, E.; Castiñeiras, A.; Domínguez, A.R.; Strahle, J.; Hiller, W. Acta Crystallogr. C, 1993, 49, 1918-1920. [ Links ]
41. Cotton, F. A.; Wilkinson, G., Advanced Inorganic Chemistry, John Wiley and Sons 5th ed., New York, 1988. [ Links ]
42. Huheey, J. E.; Keiter, E. A.; Keiter, R. L., Inorganic Chemistry. 4th ed., Harper Collins College Editions, New York, 1999. [ Links ]
43. Lever A.B. P., Inorg. Chem., 1990, 29, 1271-1285. [ Links ]
44. Stranger, R; McMahon, K.L; Gahan, L.R.; Bruce, J.I.; Hambley, T.W. Inorg. Chem. 1997, 36, 3466-3475. [ Links ]
45. D. Sutton, Electronic Spectra of Transition Metal Complexes. McGraw Hill, 1968, London, Great Britain. [ Links ]
46. Bard, A.J.; Faulkner, L.R., Electrochemical Methods, Fundamentals and Applications. 2nd ed, John Wiley and Sons, New York, 2001. [ Links ]
47. Kissinger, P. T. and Heineman, W. R., Laboratory Techniques in Electroanalytical Chemistry, Marcel Dekker, Inc. New York, USA, 1996. [ Links ]
48. Zanello, P., Inorganic Electrochemistry, theory, practice and application, The Royal Society of Chemistry, Cambridge, UK, 2003. [ Links ]
49. Lappin A. Graham; McAuley Alexander The redox chemistry of Nickel, 281-295. [ Links ]
50. Tokel-Takvoryan, N.E.; Hemingway, R. E.; Bard, A. J., J. Am. Chem. Soc., 1973, 95, 6582-6589. [ Links ]
51. Tanaka, N.; Sato, Y.; Bull. Chem. Soc. Jap. ,1968, 41, 2059-2064. [ Links ]
52. Margel, S.; Smith, W.; Anson, F.C. J. Electrochem. Soc., 1978, 125, 241-246. [ Links ]
53. Tanaka, N.; Sato, Y., Inorg. Nucl. Chem. Lett., 1968, 4, 487-490. [ Links ]
54. Tanaka, N.; Sato, Y., Inorg. Nucl. Chem. Lett., 1966, 2, 359-362. [ Links ]
55. Tanaka, N.; Sato, Y., Electrochim. Acta.,1968, 335-346. [ Links ]
56. Christensen P.A. ; Hamnett A.; Higgins S.J.; Timney, J. A., J. Electroanal. Chem., 1995, 395, 195-209. [ Links ]
57. Salvatore, D.; Paolo, U., J. Electroanal. Chem., 1987, 219, 259-271. [ Links ]
58. Nicholson, R. S., Anal. Chem. 1965, 37, 1351-1355. [ Links ]
59. Perpone, S.P.; Kretlow, W.J.,Anal. Chem. 1966, 38, 1760-1763. [ Links ]
60. Solomon, E.I.; Lever A.P.B., Inorganic Electronic Structure and Spectroscopy, Volume II: Applications and Case Studies, Wiley, New York, 1999, 255-262. [ Links ]
61. Gritzner, G. and Küta J., Pure Appl. Chem, 1984, 4, 461-466. [ Links ]

