










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkJournal of the Mexican Chemical Society
Print version ISSN 1870-249X
J. Mex. Chem. Soc vol.58 n.3 Ciudad de México Jul./Sep. 2014
Article
Electrochemical Behavior of Metamitron Herbicide at the Interface of Two Immiscible Electrolyte Solutions
Alma Gricel Reyes-Reyes,a Judith Amador-Hernández,b and Miguel Velázquez-Manzanaresa*
a Institute of Biotechnology, Universidad del Papaloapan, Circuito Central 200, 68301 San Juan Bautista Tuxtepec, Oaxaca, Mexico. mvmiguel@hotmail.com
b Faculty of Sciences, Universidad Autónoma de Coahuila, Blvd. V. Carranza S/N, 25280 Saltillo, Coahuila, México.
Received February 5th, 2014
Accepted April 3rd, 2014.
Abstract
The electrochemical behavior of the metamitron herbicide at the water|1,2-dichloroethane interface was studied by means of cyclic voltammetry and Electrochemical Impedance Spectroscopy. The results show that metamitron extraction across the interface was pH dependent. The curves capacitance-potential shown that the presence of a lipid monolayer inhibits the metamitrone transport across the interface. In this paper the equilibrium partition of metamitron across the interface of two immiscible electrolyte solutions is discussed.
Key words: Liquid|liquid interface, ITIES, metamitron, herbicides, capacitance-potential curves.
Resumen
Se estudió el comportamiento electroquímico del herbicida metamitrona en la interface agua|1,2-dicloroetano mediante voltamperometría cíclica y Espectroscopía de Impedancia Electroquímica. Se encontró que la extracción de la metamitrona es dependiente del pH. Los estudios a través de las curvas capacitancia-potencial muestran que la presencia de una monocapa de lípidos en la interfase, bloquea el transporte de este herbicida. En el presente articulo se discute el equilibrio de partición de la metamitrona en el sistema de interfases de dos soluciones electrolíticas inmiscibles.
Palabras clave: Interfase líquido|líquido, IDSEI, metamitrona, herbicidas, curvas capacitancia-potencial.
Introduction
Ion and drug transfer across biological membranes is an important issue that has been addressed by several research projects [1,2]. Currently, the study of the partition of herbicides through biphasic systems has become very important from an environmental point of view, for example, the bioaccumulation that these compounds may have on living systems.
Metamitron (MET) is a synthetic compound that belongs to the family of triazine herbicides. This compound is selective in eliminating unwanted plants in agriculture and can be used on pre-emergence or post-emergence weeds. In fact, this chemical has been used frequently in recent years, since it ensures commercially important crops. At the molecular level, the mechanism how this herbicides acts is based on the disruption of the photosystem II in the plant leaf, which basically inhibits the electron transfer process. This provokes the destruction of chlorophyll and carotenoids, causing chlorosis and the free radical formation that destroys membrane cells [3].
The main chemical structure of triazine herbicides is a ring of six members, with three nitrogen and three carbon atoms. The properties of symmetric triazines are marked by the substituent in position 2 of the ring, which can be a chloride (Cl), metoxy (OCH3) or thiometyl (SCH3) group. The substituents in positions 4 and 6 on the ring are alkyl-amine groups that increase the hydrophobicity of this kind of molecules [4, 5].
Metamitron (MET), or 4-amine-3-dihydro-3-methyl-6-phenyl-1,2,4-triazine-5-ona according to IUPAC, is classified as an asymmetric herbicide. It is widely used for the control of grasses and broad-leaved weeds in sugar and red beets, fodder beet, and certain strawberry varieties. MET is slightly toxic through ingestion, with reported oral LD50 values of 3343 mg kg−1 in rats [5].This chemical is a weak base (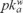 = 1.9). Due to its high water solubility, it can be highly mobile when applied to soils and consequently can become an environmental contaminant of surface and ground waters. It degrades in soils mainly through microbial transformation, although its photolysis by sunlight has also been reported [6].
= 1.9). Due to its high water solubility, it can be highly mobile when applied to soils and consequently can become an environmental contaminant of surface and ground waters. It degrades in soils mainly through microbial transformation, although its photolysis by sunlight has also been reported [6].
The Interface of Two Immiscible Electrolyte Solutions (ITIES) has been an interesting tool in studying the partition equilibrium of ionic species across hydrophobic barriers [7-9].
The ITIES can be useful in simplifying the ion transfer mechanism across biological membranes. Professor Koryta was the first to postulate that the ITIES could behave as a metal electrode|electrolyte solution interface [10], so that traditional electrochemical techniques such as cyclic voltammetry and ac impedance spectroscopy can be applied to the study of the charge transfer across the ITIES [11].
There are several studies dealing with the organic molecule transfer across ITIES, such as those focusing on peroxicam and quinidine, which have attempted to determine the mechanism for drug partition across hydrophobic barriers; [12, 13]. In these cases, thermodynamic studies were presented for the assisted proton transfer at the ITIES where the authors constructed ionic partition diagrams of ionisable compounds as a function of pH and Galvani interfacial potential (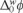 ). This latter provides information on ionic transfer behavior; that is, whether it is a simple ionic transfer or a facilitated proton transfer by ionisable drugs. The ITIES has also been a tool for studying small organic molecules as a product of antibiotic degradation, which had to be preconcentrated and quantified by another analytical method, this was possible since these small molecules remain in ionic form [14]. More recently, the transfer of herbicides of the triazine families has also been studied [15], as well as the competitive transfer between H+ and Al3+ by the herbicide prometryn at the ITIES [16]. The ITIES has also been applied to study targets in biological samples, such as the electrochemical behavior of peptide mixtures resulting from enzymatic protein digestion [17], or the extraction of propranolol and timolol from artificial urine [18].
). This latter provides information on ionic transfer behavior; that is, whether it is a simple ionic transfer or a facilitated proton transfer by ionisable drugs. The ITIES has also been a tool for studying small organic molecules as a product of antibiotic degradation, which had to be preconcentrated and quantified by another analytical method, this was possible since these small molecules remain in ionic form [14]. More recently, the transfer of herbicides of the triazine families has also been studied [15], as well as the competitive transfer between H+ and Al3+ by the herbicide prometryn at the ITIES [16]. The ITIES has also been applied to study targets in biological samples, such as the electrochemical behavior of peptide mixtures resulting from enzymatic protein digestion [17], or the extraction of propranolol and timolol from artificial urine [18].
There is an increasing interest in the study of biomolecules at the ITIES, since the ITIES can be used as a good approach to mimicking the membrane|water interface when the interface is modified with lipids. The adsorption of phospholipids of different long chains at the water|1,2-dichloroethane interface has been studied through Electrochemical Impedance Spectroscopy (EIS), such as distearoyl-phosphatidylcholine (DSPC), dioleyl-phosphatidylcholine (DOPC), dilauril-phosphatidylcholine (DLPC), dimiristoyl-phosphatidylcholine (DMPC), or dipalmitoyl-phosphatidylcholine (DPPC) . Those studies showed that the lipid concentration has an effect on the ionic partitioning process; in fact, a compact monolayer of lipids may likely form when the lipid concentration increases [19]. Another interesting example is the study of the transfer of the acetylcholine across the water|1,2-DCE interface modified with dipalmitoyl-phosphatidylcholine (DPPC); in this case, the apparent rate constant for the acetylcholine transfer was obtained by EIS, and it was found that the presence of the DPPC monolayer decreases the apparent rate constant of this compound [20].
The aim of this work was to study the electrochemical behavior of MET across the water|1,2-dichloroethane interface in order to provide information to help recognize their behavior in the environment and particularly in exposed organisms.
Results and discussions
Cyclic voltammetric studies
Initially, MET (Fig. 1) was placed into the organic phase for twenty minutes before the CV was performed. The experiments were carried out at different pH values, and a few representative voltammograms are shown in Figure 2. All voltammogram show one reversible wave in the whole range of pH studied with a peak-to-peak separation of 60 mV, which means that a single charge is transferred across the interface, as was expected by the theoretical approach given by the Nernst equation (59 mV).
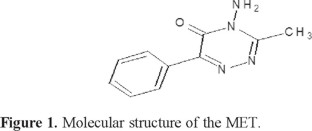
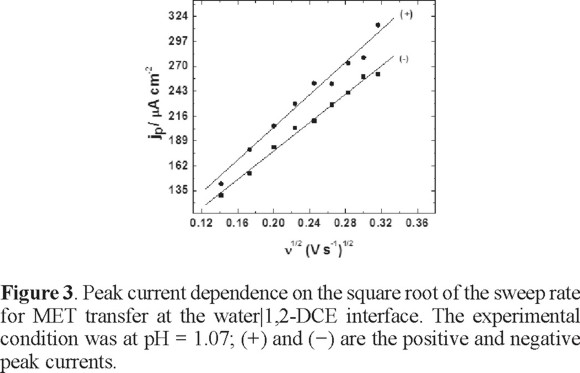
MET has a  = 1.9 and the protonation takes place at the nitrogen atom of the aromatic ring [21]. The CV performed at pH= 1.07 indicates that MET is transferred as a whole protonated molecule (Figure 2); the transfer mechanism will be discussed later. A linear dependence between the current peak with the square root of sweep rate potential was observed, meaning that the transfer is fast and controlled by diffusion of the protonated form of the compound (Figure 3). This electrochemical behavior was similar for the whole range of pH that was studied. In the CV, the current signal attributable to the ionic transfer begins at pH 4.5; at this pH, it is not difficult to measure the half-wave potential. However, at pH higher than 5.0, the half-wave potential was difficult to measure since the supporting electrolyte was also transferring at large positive potentials. Similar electrochemical behaviour has been observed for other compounds [15]. Thus, when the pH becomes more acidic, the half-wave potential shifts to the centre of the potential window; due to the fact that MET is easier to extract as the proton concentration increases.
= 1.9 and the protonation takes place at the nitrogen atom of the aromatic ring [21]. The CV performed at pH= 1.07 indicates that MET is transferred as a whole protonated molecule (Figure 2); the transfer mechanism will be discussed later. A linear dependence between the current peak with the square root of sweep rate potential was observed, meaning that the transfer is fast and controlled by diffusion of the protonated form of the compound (Figure 3). This electrochemical behavior was similar for the whole range of pH that was studied. In the CV, the current signal attributable to the ionic transfer begins at pH 4.5; at this pH, it is not difficult to measure the half-wave potential. However, at pH higher than 5.0, the half-wave potential was difficult to measure since the supporting electrolyte was also transferring at large positive potentials. Similar electrochemical behaviour has been observed for other compounds [15]. Thus, when the pH becomes more acidic, the half-wave potential shifts to the centre of the potential window; due to the fact that MET is easier to extract as the proton concentration increases.
In order to know the hydrophobic nature of this herbicide, the formal Gibbs energy of ion transfer of ion i,  , is given by:
, is given by:

where  is the formal Galvani ionic transfer potential for ion i, zi is its charge and F is the Faraday constant.
is the formal Galvani ionic transfer potential for ion i, zi is its charge and F is the Faraday constant. 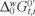 gives a measure of the hydrophobicity of MET and can be related to its chemical structure. The measured half-wave potential E1/2 was converted to the Galvani potential scale and
gives a measure of the hydrophobicity of MET and can be related to its chemical structure. The measured half-wave potential E1/2 was converted to the Galvani potential scale and 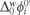 was calculated from [22]:
was calculated from [22]:
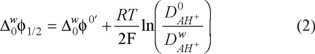
 includes the activity coefficient of the proton in the aqueous phase and the protonation equilibrium constant. The ratio Do/Dw was evaluated from Walden's rule Do/Dw = ηw/ηo, where ηo and ηw are the viscosity coefficients of the organic solvent and water, respectively, thus the value for the Do/Dw = 1.02, this values was used to evaluate the
includes the activity coefficient of the proton in the aqueous phase and the protonation equilibrium constant. The ratio Do/Dw was evaluated from Walden's rule Do/Dw = ηw/ηo, where ηo and ηw are the viscosity coefficients of the organic solvent and water, respectively, thus the value for the Do/Dw = 1.02, this values was used to evaluate the 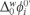 . The physicochemical parameters from the CV, such as the
. The physicochemical parameters from the CV, such as the 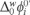 = 0.21 V, was calculated using equation (2). From this value, was found a value of the
= 0.21 V, was calculated using equation (2). From this value, was found a value of the  = 20.26 kJ mol−1. These values were estimated considering the acid conditions lower than
= 20.26 kJ mol−1. These values were estimated considering the acid conditions lower than  , where the molecule was completely protonated. In a previous work, the Gibbs energy for a s-triazine with a methyl group (-CH3) in position 2 was found to be -2.6 kJmol−1 [23] . On the other hand, the difference in the Gibbs energy for the halogenated s-triazines named atrazine (20.6 kJmol−1) and simazine (21.0 kJmol−1) [15] can be attributed to the presence of the -CH3 group (atrazine) instead of H (simazine) in an alquil-amine substituent of the triazine ring. In the case of MET, the presence of the aromatic ring on its chemical structure should make the molecule more hydrophobic; however, that was not the case, since the
, where the molecule was completely protonated. In a previous work, the Gibbs energy for a s-triazine with a methyl group (-CH3) in position 2 was found to be -2.6 kJmol−1 [23] . On the other hand, the difference in the Gibbs energy for the halogenated s-triazines named atrazine (20.6 kJmol−1) and simazine (21.0 kJmol−1) [15] can be attributed to the presence of the -CH3 group (atrazine) instead of H (simazine) in an alquil-amine substituent of the triazine ring. In the case of MET, the presence of the aromatic ring on its chemical structure should make the molecule more hydrophobic; however, that was not the case, since the  is similar to atrazine and simazine. The explanation for this behavior is that MET can easily interact with water through hydrogen bonds at its carboxylic and amino substituents, since the triazine ring is less protected by the hydrophobic groups.
is similar to atrazine and simazine. The explanation for this behavior is that MET can easily interact with water through hydrogen bonds at its carboxylic and amino substituents, since the triazine ring is less protected by the hydrophobic groups.
The ionic partition diagram
The ion partition diagram was constructed from the CV experiments, which were carried out at different pH values of the aqueous phase and at a constant MET concentration (see Figure 4). For pH <  in the range of pH between 2 to 1.8, the thermodynamic equilibrium was displaced sufficiently to induce a simple ion transfer where the whole molecule was transferred. In this case, the predominant specie is the protonated form (METH+), as has been mentioned before. The difference of Galvani potential is independent of pH, as is shown in the Figure 4. MET transfer can be described according to:
in the range of pH between 2 to 1.8, the thermodynamic equilibrium was displaced sufficiently to induce a simple ion transfer where the whole molecule was transferred. In this case, the predominant specie is the protonated form (METH+), as has been mentioned before. The difference of Galvani potential is independent of pH, as is shown in the Figure 4. MET transfer can be described according to:
METH+ (w) → METH+ (o) (I)
where METH+(w) and METH+(o) are the protonated molecule in the aqueous and organic phase, respectively. When the experimental conditions are in the range of pH values between 3.0 and 5.0 (pH >  ) (Figure 4), the potential becomes dependent of the pH, as can be observed in the pH-potential diagram. It is probable that MET remains in the organic phase and acts as a proton carrier at the interface; a similar behavior has been observed in other studies [12, 13, 15]. In order to prove that mentioned before, the interface was modified with different concentrations of α-phophatidylcholine (PC).
) (Figure 4), the potential becomes dependent of the pH, as can be observed in the pH-potential diagram. It is probable that MET remains in the organic phase and acts as a proton carrier at the interface; a similar behavior has been observed in other studies [12, 13, 15]. In order to prove that mentioned before, the interface was modified with different concentrations of α-phophatidylcholine (PC).
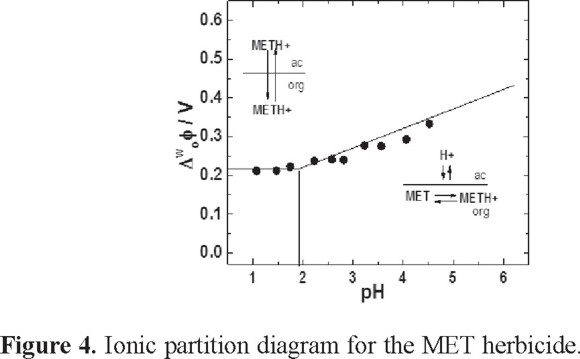
The experiments with PC were performed at pH values of 1.03 and 3.06, where the electrochemical behavior of MET can be clearly observed. Figure 5 shows capacitance-potential curve series for different concentrations of PC at both pH conditions. As can be seen at pH = 3.06, the largest capacitance values corresponded to the baseline. When PC was injected into the organic phase, the capacitance decreased significantly, due to the PC starts to form a monolayer at the interface, as has been observed in previous studies [20]. Definitely, the lipid monolayer behavior depends on the applied potential; at 25 and 30 µM of PC, the capacitance decreases significantly as soon as the equilibrium is reached, which probably occurred when the PC was completely packed at the interface. At pH = 1.03 the capacitance values are larger than values observed at pH = 3.06. Under this condition, the capacitance values decrease as a function of the PC concentration. Despite the excess of proton concentration, the formation of the monolayer is achieved at high lipid concentrations. The large values of the capacitance is due to the excess of protons in the vicinity of the interface, which greatly increases the surface charges, and hence the capacitance. The capacitance values are in the order of magnitude of 1.7-1.9 µF cm−2, similar to the observed values in studies of a lipid monolayer on mercury electrodes [24].
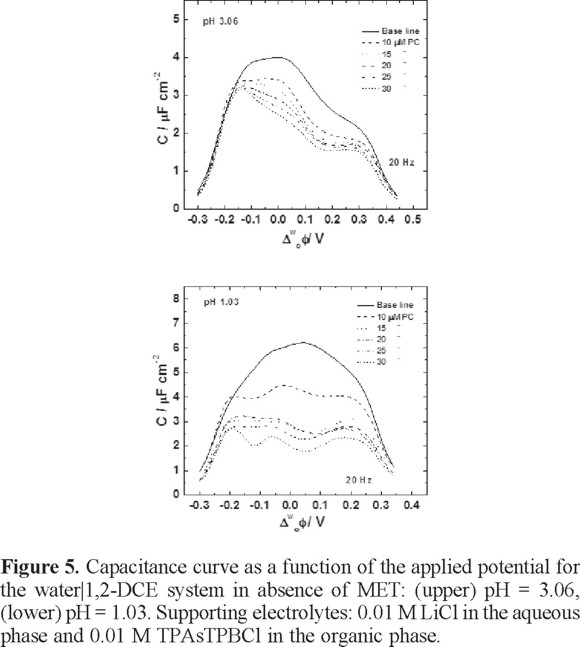
Figure 6 shows the capacitance curves obtained in presence of MET and at different PC concentrations. The capacitance values decreased when MET is present, as can be observed. At pH = 3.06, the capacitance values around 0.2-0.3 V indicate the transfer of MET across the interface in presence of the PC monolayer. Although the MET is able to transfer across the interface, the capacitance perhaps depends of the diffusion of the MET in the organic phase, since at this pH the herbicide acts as a proton carrier and the protonation takes place at the interface according to the ionic partition diagram. This behavior can be observed more clearly in a CV for the MET transfer when the interface is modified with PC (figure 7), as can be observed, the current peak increases as a function of PC concentrations meaning that diffusion process of the MET are modified in presence of the PC at the interface. It is true that the presence of the PC in some way inhibits the transfer of the herbicide. In previous studies on this matter, an ionophore named Antibiotic 23187 has been incorporated into a phospholipids monolayer on mercury electrodes in order to study the effect of this kind of compound on the permeability of the monolayer [25]. As a result, the presence of a lipid monolayer inhibits the oxidation of metal ions on the metal electrode, but the presence of the ligand in the monolayer makes the interaction of the metal ion with the electrode favorable.
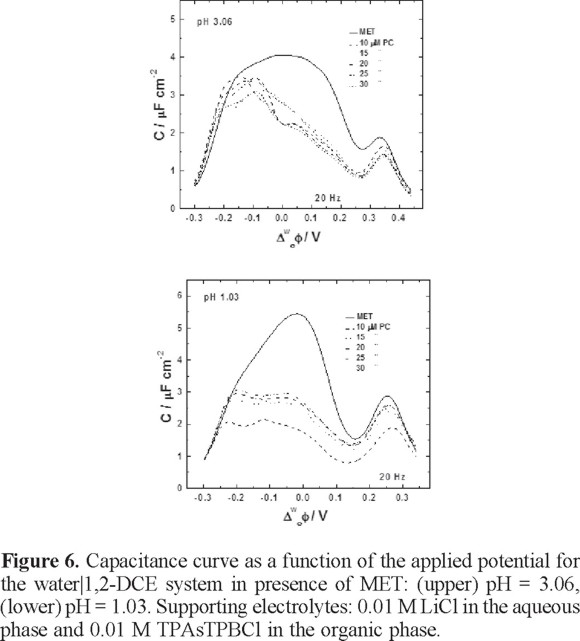
On the other hand, at pH = 1.03, the capacitance values decreased in a more pronounced way between 0.1-0.2 V. At this pH, MET is in ionic form, therefore is completely transferred through the interface as a consequence of the abrupt interruption of the lipid monolayer.
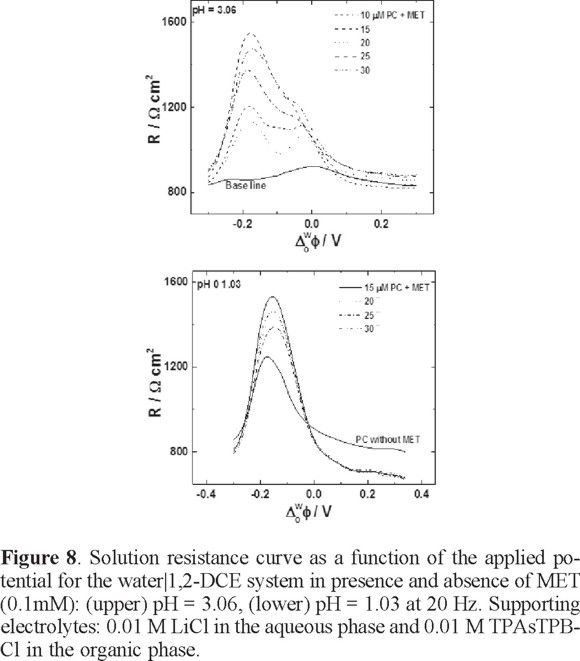
In figure 8, the resistance of the solution is increased as a function of the PC concentration compared with the signal in absence of MET. This is logical because at better organization of the PC monolayer, the resistance is higher. The presence of MET in the system significantly reduces the resistance; of course, this is observed in the potential region where the MET is transferred. These results are consistent with the cyclic voltammetry and potential curve experiments.
Conclusions
It has been demonstrated that the transfer of MET across the interface of two immiscible electrolyte solutions depends on the pH of the aqueous phase. At pH <  , the herbicide is transferred across the interface in its ionic form, while at pH >
, the herbicide is transferred across the interface in its ionic form, while at pH >  this molecule can facilitate proton transfer across hydrophobic barriers. The presence of the lipid monolayer is enable the blocking the transfer of MET across the interface, although the transfer mechanism is still pH dependent. These results provide valuable information about the behavior of this kind of contaminants across the lipid barriers of biological membranes.
this molecule can facilitate proton transfer across hydrophobic barriers. The presence of the lipid monolayer is enable the blocking the transfer of MET across the interface, although the transfer mechanism is still pH dependent. These results provide valuable information about the behavior of this kind of contaminants across the lipid barriers of biological membranes.
Experimental part
The cyclic voltammetry was carried out in a glass cell with a four electrode configuration, with a contact area between the two immiscible liquids of 0.2 cm2. The interfacial potential was controlled with a potenciostat/galvanostat (Gamry, Reference 600, USA). The measured potential corresponds to the following cell:

where x mM is the MET concentration in the organic phase; σ represents the interface of study, and SCE1 and SCE2 are saturate calomel electrodes. A semi-micro pH electrode coupled to a pHmeter (ThermoOrion, USA), was used to measure pH directly in the electrochemical cell.
Tetraphenylarsonium chloride (TPAsCl, Fluka), 1,2-dichloroethane (1,2-DCE, Gold Label, Aldrich) and potassiumtetrakis(4-chlorophenyl)borate (KTPBCl, Fluka purum) were used as received. Tetraphenylarsonium tetrakis(4-chlorophenyl)borate (TPAsTPBCl) was formed from the corresponding chemicals and re-crystallized twice from acetone (BDH, AnalaR). MET was pestanal grade (Sigma-Aldrich). LiCl (Sigma-Aldrich) was used as supporting electrolyte in the aqueous phase. The pH was adjusted with H2SO4 and LiOH (Sigma-Aldrich) and the aqueous solutions were prepared in ultrapure water (Easypure UV, Barnstead).
All the experiments were carried out at 25 ± 1 oC. During measurements, the electrochemical cell was placed inside a Faraday cage. The interface was modified with L-α-phophatidylcholine (Sigma) and the capacitance studies were carried out by EIS and at single frequency (20 Hz) with the Mott-Schottky technique. Experimental data were simulated using an electronic circuit consisting of a capacitor in series with a resistor.
The potentials were reported on the Galvani potential scale and calculated according to [26]:
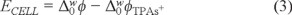
where  is the Galvani interfacial potential and
is the Galvani interfacial potential and  the organic reference liquid junction potential [23]. The standard transfer potential of TPAs+,
the organic reference liquid junction potential [23]. The standard transfer potential of TPAs+,  for TPAs+ was taken as -0.364 V [27].
for TPAs+ was taken as -0.364 V [27].
Acknowledgments
Consejo Nacional de Ciencia y Tecnología (CONACyT, México) is gratefully acknowledged by the financial support of this work (Project CB-2010/152918).
Reference
1. Loftsson, T.; Vogensen, S. B.,; Brewster, M. E.; Konráosdóttir, F. J. Pharm. Sci. 2007, 96, 2532-2546. [ Links ]
2. Kansy, M.; Senner, F.; Gubernator, K. J. Med. Chem. 1998, 41, 1007-1010. [ Links ]
3. Duke S. O.; Dayan, F. E. In: Uso de Herbicidas en la Agricultura del Siglo XXI. De Prado, R; Jorrín J. V., Ed. Universidad de Córdoba, Córdoba Spain, 2001, 31-44. [ Links ]
4. LeBaron, H. M.; McFarland, J. E.; Burnside, O. C., Eds. The Triazine Herbicides: 50 years Revolutionizing Agriculture. Elsevier, Amsterdam, 2008. [ Links ]
5. Budavari, S.; O'Neil, M.; Smith, A.; Heckelman, P.; Obenchain, J. The Merck Index, An Encyclopedia of Chemicals, Drugs and Biologicals 12th ed. CRC Press, Boca Raton FL, 1996. [ Links ]
6. Palm, W. U.; Millet, M.; Zetzsch, C. Chemosphere 1997, 35, 1117-1130. [ Links ]
7. Peljo, P,; Girault, H. H. Encyclopedia of Analytical Chemistry. Wiley, New York, 2012. [ Links ]
8. Peljo, P.; Rauhala, T.; Murtomaki, L.; Kallio, T.; Kontturi, K. Int. J. Hydrogen Energy 2011, 36, 10033-10043. [ Links ]
9. Sanchez, V. L. J.; Ovejero, J. M.; Fernández, R. A.; Dassie, S. A. Int. J. Electrochem. 2012, 2012, 1-34. [ Links ]
10. Koryta, J.; Vanysek, P.; Brezina, M. J. Electroanal. Chem. Interfac. Electrochem. 1976, 67, 263-266. [ Links ]
11. Itagaki, M.; Fukushima, H.; Inoue, H.; Watanabe, K. J. Electroanal. Chem. 2001, 504, 96-103. [ Links ]
12. Reymond, F.; Steyaert, G.; Pagliara, A.; Carrupt, P-A.; Testa, B.; Girault, H. Helv. Chim. Acta 1996, 79, 1651-1669. [ Links ]
13. Reymond, F.; Steyaert, G.; Carrupt, P-A.; Testa, B.; Girault, H. Helv. Chim. Acta, 1996, 79, 101-117. [ Links ]
14. Vanysek, P.; Basáez, L. J. Pharm. Biomed. Anal. 1999, 19, 183-192. [ Links ]
15. Velázquez-Manzanares, M.; Amador-Hernández, J.; Cisneros-Cisneros, C.; Heredia-Lezama, K. J. Electrochem. Soc. 2008, 155, F218-F222. [ Links ]
16. Juarez, A.V.; Yudi, L. M. Electrochim. Acta. 2008, 54, 530-534. [ Links ]
17. Herzog, G.; Roger A.; Sheehan, D.; Arrigan, D. W. Anal. Chem. 2010, 82, 258-264. [ Links ]
18. Collins, C. J.; Berduque, A; Arrigan, D. W., Anal Chem. 2008, 80, 8102-8108. [ Links ]
19. Martins, M. C.; Pereira, C. M.; Santos, H. A.; Dabirian, R.; Silva, F.; Garcia-Morales, V.; Manzanares, J. A. J. Electroanal. Chem. 2007, 599, 367-375. [ Links ]
20. Santo, H. A.; Pereira, C. M.; Silva, F. Portugaliae Electrochim. Acta. 2005, 22,263-274. [ Links ]
21. Pinto, G. M.; Jardim, I. C. J. Chromat. A 2000, 869, 463-469. [ Links ]
22. Homolka, D.; Marecek, V.; Samec, Z.; Base, K.; Wendt, H. J. Electroanal. Chem. 1984, 163, 159-170. [ Links ]
23. Cunnane, V. J.; Schiffrin, D. J.; Beltran, C.; Geblewicz, G.; Solomon, T. J. Electroanal. Chem. Interfac. Electrochem. 1988, 247, 203-214. [ Links ]
24. Moncelli, M. R.; Becucci, L.; Guidelli, R. Biophys. J. 1994, 66, 1969-1980. [ Links ]
25. Nelson, A. J. Chem. Soc. Faraday. Trans. 1991, 87, 1851-1856. [ Links ]
26. Wiles, M. C.; VanderNooot, T. J.; Schiffrin, D. J. J. Electroanal. Chem. 1999, 281, 231-244. [ Links ]
27.Koryta, J.; Vanysek, P.; Brezina, M.; J. Electroanal. Chem. Interfac. Electrochem. 1977, 75, 211-228. [ Links ]

