










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkJournal of the Mexican Chemical Society
Print version ISSN 1870-249X
J. Mex. Chem. Soc vol.58 n.3 Ciudad de México Jul./Sep. 2014
Review
Electro-Fenton, UVA Photoelectro-Fenton and Solar Photoelectro-Fenton Treatments of Organics in Waters Using a Boron-Doped Diamond Anode: A Review
Enric Brillas*
Laboratori d'Electroquímica dels Materials i del Medi Ambient (LEMMA), Departament de Química Física, Facultat de Química, Universitat de Barcelona, Martí i Franquès 1-11, 08028 Barcelona, Spain. brillas@ub.edu
Received January 10th, 2014
Accepted March 11th, 2014.
Abstract
The characteristics and applications of electrochemical advanced oxidation processes (EAOPs) like electro-Fenton (EF), UVA photoelectro-Fenton (PEF) and solar PEF (SPEF) with a boron-doped diamond (BDD) anode are reviewed. Their oxidative properties are based on the attack of •OH formed at the anode surface and in the medium from Fenton's reaction between cathodically generated H2O2 and added Fe2+. The illumination with UVA light in PEF and sunlight in SPEF enhances the degradation process due to the photolysis of complexes of Fe(III) with generated carboxylic acids. Examples on the removal of industrial chemicals, pesticides, dyes and pharmaceuticals by these EAOPs using bench-scaled stirred tank reactors and pre-pilot plants are described. The influence of experimental variables on the mineralization and energetic parameters is detailed. The decay kinetics of pollutants and the evolution of intermediates are discussed. The SPEF process shows the best performance, being the most potent EAOP tested.
Keywords: Electro-Fenton, mineralization, oxidation products; photoelectro-Fenton, sunlight, UVA light, water treatment.
Resumen
Este artículo revisa las características y aplicaciones de procesos electroquímicos avanzados de oxidación (PEAOs) como el electro-Fenton (EF), el fotoelectro-Fenton UVA (FEF) y el FEF solar (FEFS) con un ánodo de diamante dopado con boro (DDB). Sus propiedades oxidativas se basan en el ataque del radical •OH formado en la superficie del ánodo y en el medio a partir de la reacción de Fenton entre el H2O2 generado catódicamente y el Fe2+ añadido. La iluminación con luz UVA en PEF y luz solar en FEFS mejora el proceso degradativo debido a la fotólisis de complejos de Fe(III) con ácidos carboxílicos generados. Se describen ejemplos sobre la eliminación de productos químicos industriales, pesticidas, colorantes y fármacos por estos PEAOs usando reactores de tanque agitado a pequeña escala y plantas pre-piloto. También se detalla la influencia de variables experimentales sobre los parámetros de mineralización y energéticos y se discute el descenso cinético de los contaminantes y la evolución de sus intermedios. El proceso de FEFS resulta ser el más eficiente, siendo el más potente PEAO ensayado.
Palabras clave: Electro-Fenton, mineralización, productos de oxidación, fotoelectro-Fenton, luz solar, luz UVA, tratamiento de aguas.
Introduction
A high number of synthetic organics like industrial chemicals, pesticides, dyes and pharmaceuticals are released daily into large volumes of wastewaters, thus entering into natural waters where they accumulate in the aquatic environment [1-4]. This contamination proceeds from urban, industrial and agricultural human activities and cannot be significantly removed by conventional wastewater treatment plants because most synthetic organics are recalcitrant, highly stable to sunlight irradiation and with large resistance to microbial attack and temperature. Low amounts of many synthetic organics, usually at µg L−1 level, have been detected in rivers, lakes, oceans and even drinking water all over the world. Since water is essential for the subsistence of all the living beings, this pollution remains a pervasive threat. Research efforts are being made to remove organics from waters and wastewaters using safe, effective and economical technologies in order to avoid their toxic consequences and potential hazardous health effects on the living beings.
Over the past two decades, many advanced oxidation processes (AOPs) have attracted increasing interest for the efficient destruction of toxic and/or biorefractory pollutants from waters [2, 5-10]. AOPs are powerful environmental friendly technologies involving chemical, photocatalytic, photochemical, electrochemical and photoelectrochemical methods in which hydroxyl radical (•OH) is produced in situ as the main oxidant. The high standard reduction potential (Eº(•OH/H2O) = 2.80 V vs. SHE) of this radical ensures that it can react non-selectively with most organics yielding dehydrogenated or hydroxylated derivatives, which can be in turn mineralized to CO2, water and inorganic ions [8-10]. The most typical chemical AOP is the Fenton method based on the use of a mixture of Fe2+ and H2O2 (Fenton's reagent) to destroy organics. The oxidation ability of this method can be significantly enhanced by irradiating the treated effluent with UV light (photo-Fenton method) or sunlight (solar photo-Fenton method) [9]. Another way to enhance the decontamination efficiency of the Fenton process is its coupling with electrochemical methods, giving rise to a large variety of electrochemical AOPs (EAOPs) [7, 8]. They present many technical advantages such as environmental compatibility, versatility, high efficiency, amenability of automation and safety because they operate at mild conditions [2]. EAOPs are mediated electrochemical treatments based on the destruction of organics at the anode and/or using the Fenton's reagent partially or completely generated from electrode reactions.
The simplest EAOP based on the destruction of organics at the anode is the electro-oxidation or anodic oxidation (AO) method. When it is also carried out with cathodic H2O2 generation, it is so-called AO with electrogenerated H2O2 (AO-H2O2) [7]. If Fenton's reagent is electrogenerated, the EAOPs based on Fenton's reaction chemistry are considered, which have been developed for the treatment of acidic wastewaters [2, 8, 9]. The most typical technique is the electro-Fenton (EF) process, where an iron catalyst is added to the effluent while H2O2 is produced at the cathode with O2 or air feeding. The oxidation ability of the EF method depends on the anode material and it has been found that boron-doped diamond (BDD) electrodes yield the best performance, as demonstrated firstly in our laboratory in 2004. The efficiency of EF can also be improved by combining it with the simultaneous illumination of the solution by UVA light or sunlight, corresponding to the so-called UVA photoelectro-Fenton (PEF) and solar PEF (SPEF) methods [8].
This paper aims to present a general review over the application of the EF, PEF and SPEF methods to the degradation of organic pollutants in waters using potent BDD anodes. Examples on the treatments of industrial chemicals, pesticides, dyes and pharmaceuticals are examined to show the high oxidation ability of these EAOPs. Fundamentals of these methods are initially described to better analyze their characteristics and oxidative properties.
Fundamentals of the EF, PEF and SPEF Methods
Hydrogen peroxide is a "green" chemical since it leaves oxygen gas and water as byproducts. This product is used to clean electronic circuits, delignify agricultural wastes and bleach pulp and paper and textiles, for disinfection in medical and industrial applications and as an oxidant in synthesis and wastewater treatment [11]. However, H2O2 can only attack reduced sulfur compounds, cyanides and certain organics such as aldehydes, formic acid and some nitro-organic and sulfo-organic compounds. The treatment of wastewaters with H2O2 is then limited by its low oxidation ability and for this reason, it is activated in acidic effluents with Fe2+ ion as catalyst yielding the Fenton's reagent to produce homogenous •OH as strong oxidant of organics in the well-known chemical Fenton method [8].
It is known since 1882 that H2O2 can be produced in aqueous media from the two-electron reduction of dissolved O2 gas at carbonaceous cathodes with high surface area [12]. In acidic medium, this reaction with Eº = 0.68 V vs. SHE can be written as follows:
O2(g) + 2 H+ + 2 e− → H2O2 (1)
Reaction (1) is easier than the four-electron reduction of O2 to water (Eº = 1.23 V vs. SHE). Electrochemical reduction at the cathode surface from reaction (2) and, in much lesser extent, disproportion in the bulk from reaction (3) are parasitic reactions that result in the loss of oxidant or a lowering of current efficiency [13]:
H2O2 + 2 e− → 2 OH− (2)
2 H2O2 → O2(g) + 2 H2O (3)
In an undivided cell, H2O2 is also anodically oxidized to O2 via hydroperoxyl radical (HO2•) as intermediate, a weaker oxidant than •OH, by the following reaction [8,10]:
H2O2 → HO2• + H+ + e− (4)
The treatment of acidic aqueous solutions by EAOPs based on Fenton's reaction chemistry involves the continuous generation of H2O2 from O2 directly injected as pure gas or compressed air via reaction (1). O2 is efficiently reduced at cathodes like carbon nanotubes- polytetrafluoroethylene (PTFE) [14, 15], carbon nanotubes immobilized onto graphite plates [16], carbon felt (CF) [17, 18], activated carbon fiber [19, 20], carbon- PTFE gas diffusion electrodes (GDE) [21-24] and BDD electrodes [25]. When a small quantity of Fe2+ is added to the acidic effluent, it reacts with electrogenerated H2O2 to generate Fe3+ and •OH in the bulk from the Fenton's reaction (5) with absolute rate constant k2 = 76 M−1 s−1 and optimum pH of 2.8 [26]:
Fe2+ + H2O2 → Fe3+ + •OH + OH− (5)
Reaction (5) can be propagated by the catalytic behavior of the Fe3+/Fe2+ pair [27, 28]. Thus, Fe2+ can be regenerated by H2O2 from the Fenton-like reaction (6), by HO2• from reaction (7) and/or by the superoxide ion (O2•−) from reactions (8) and (9):
H2O2 + Fe3+ → Fe2+ + HO2• + H+ k2 = 3.1×10−3 M−1 s−1 (6)
Fe3+ + HO2• → Fe2+ + O2 + H+k2 = 2×104 M−1 s−1 (7)
Fe3+ + O2•− → Fe2+ + O2k2 = 5×107 M−1 s−1 (8)
Fe3+ + O2•− + 2 H2O → Fe2+ + 2 H2O2k2 = 1×107 M−1 s−1 (9)
The propagation reactions involve the production of HO2• by reaction (10) and superoxide anion O2•− by reaction (11), and the attack of •OH to saturated or aromatic organics yielding dehydrogenated or hydroxylated derivatives by reactions (12) or (13), respectively.
H2O2 + •OH → H2O + HO2•k2 = 3.3×107 M−1 s−1 (10)
HO2• ⇆ H+ + O2•−K = 4.8 (11)
RH + •OH → R• + H2Ok2 = 107−109 M−1 s−1 (12)
ArH + •OH → ArHOH•k2 = 108−1010 M−1 s−1 (13)
Also, several inhibition reactions upgrade the removal of reactive •OH, for example, its reaction with Fe2+ and its dimerization to H2O2 as follows:
Fe2+ + •OH → Fe3+ + OH−k2 = 4.3×108 M−1 s−1 (14)
•OH + •OH → H2O2k2 = 5.2×109 M−1 s−1 (15)
Interestingly, an advantage of the EAOPs compared to the chemical Fenton method is that Fe2+ is regenerated from Fe3+ reduction at the cathode by reaction (16), with Eº = 0.77 V vs. SHE [8], although in an undivided electrolytic cell, Fe2+ is slowly oxidized to Fe3+ by reaction (17) [28]:
Fe3+ + e− → Fe2+ (16)
Fe2+ → Fe3+ + e− (17)
In undivided electrolytic systems with a non-active BDD anode, the quicker degradation rate of organics in EF, PEF and SPEF is usually achieved at pH near 3. They are not only destroyed by reactive oxygen species (ROS), like •OH and in much smaller extent by H2O2, HO2•, etc., but also by heterogeneous BDD(•OH) produced from water oxidation by reaction (18) [29, 30]:
BDD + H2O → BDD(•OH) + H+ + e− (18)
Operating at high current within the water discharge region, reactive BDD(•OH) can mineralize completely aromatics and unsaturated compounds such as carboxylic acids because it is produced in high quantity, much greater, for example, than Pt(•OH) formed at the Pt anode [30]. The main loss of BDD(•OH) includes its oxidation to O2 gas via reaction (19) and its dimerization to H2O2 by reaction (20) due to the low adsorption ability of •OH on BDD [31, 32]. Furthermore, the high oxidation ability of BDD favors the formation of other weaker oxidants like peroxodisulfate (S2O82−) ion from the oxidation of SO42− ion present in the electrolyte by reaction (21) and ozone by reaction (22) [30-32].
2 BDD(•OH) → 2 BDD + O2(g) + 2 H+ + 2 e− (19)
2 BDD(•OH) → 2 BDD + H2O2 (20)
2 SO42− → S2O82− + 2 e− (21)
3 H2O → O3(g) + 6 H+ + 6 e− (22)
The BDD anode is currently preferred in AO and comparison with EF, PEF and SPEF can be made using the AO-H2O2 method without Fe2+ addition to the polluted water.
Apart from the generation of BDD(•OH) and •OH as the main oxidizing agents, the PEF process involves the exposition of the acidic treated effluent to UV light. Artificial lamps providing UVA (λ = 315-400 nm), UVB (λ = 285-315 nm) and/or UVC (λ < 285 nm) lights are commonly employed. The intensity and wavelength of such radiations have significant effect on the destruction rate of organic pollutants, which can be associated with: (i) a faster Fe2+ regeneration with additional •OH generation from photoreduction of Fe(OH)2+, the pre-eminent Fe3+ species at pH 2.8-3.5, according to photo-Fenton reaction (23) [8, 26] and/or (ii) the photodecarboxylation of complexes of Fe(III) with generated carboxylic acids allowing the regeneration of Fe2+ from the general reaction (24). As an example, reaction (25) shows the photolytic process for Fe(III)-oxalate complexes (Fe(C2O4)+, Fe(C2O4)2− and Fe(C2O4)33−) [33], formed as ultimate products of aromatics.
Fe(OH)2+ + hν → Fe2+ + •OH (23)
Fe(OOCR)2+ + hν → Fe2+ + CO2 + R• (24)
2 Fe(C2O4)n(3-2n) + hν → 2 Fe2+ + (2n-1) C2O42− + 2 CO2 (25)
On the other hand, the use of an energetic UVC light causes the photolysis of some organics and the generation of more •OH from the following homolytic cleavage of H2O2 [3]:
H2O2 + hν → 2 •OH (26)
It is noteworthy that the high electrical cost of the UV lamps utilized in PEF is the main drawback of this procedure in practice. To solve this problem, our laboratory has proposed the alternative use of the SPEF method, where sunlight, as inexpensive and renewable energy source with λ > 300 nm, irradiates directly the solution [34, 35]. The higher intensity of UV light supplied by solar radiation, along with the additional absorption at λ > 400 nm, e.g., for the photolysis of Fe(III)-carboxylate complexes, improve the mineralization of SPEF compared with PEF.
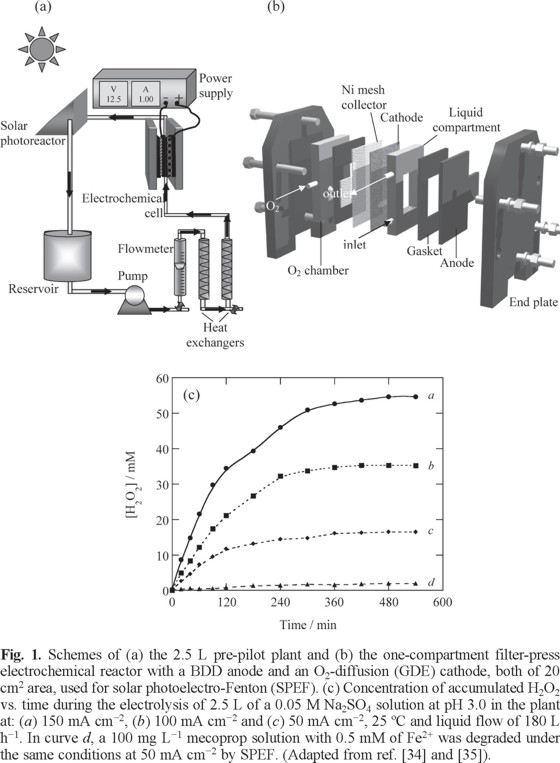
The aforementioned characteristics of the EAOPs using a BDD anode and a carbon-PTFE O2-diffusion cathode were confirmed by determining the accumulation of generated H2O2 in solution. As an example, Figs. 1a and b illustrate the scheme of a 2.5 L pre-pilot flow plant and its filter-press BDD/GDE electrochemical reactor of 20 cm2 electrode area, respectively, which have been used for the comparative study of the degradation behavior of some aromatics by AO-H2O2, EF and SPEF [34]. Fig. 1c depicts the change of H2O2 concentration during the electrolysis of 2.5 L of a 0.05 M Na2SO4 solution at pH 3.0 in the above plant at different current density [35]. In the absence of contaminants and iron ions (AO-H2O2 conditions), a gradual accumulation of H2O2 can be observed up to attain a steady concentration, which increased linearly in 17, 35 and 54 mM with rising current density in 50 mA cm−2 (curve c), 100 mA cm−2 (curve b) and 150 mA cm−2 (curve a). This trend suggests that all electrode reactions involved are faradaic and the steady H2O2 concentration was reached just when its generation rate from reaction (1) became equal to its oxidation rate at the anode from reaction (4). In contrast, when 100 mg L−1 of the herbicide mecoprop and 0.5 mM Fe2+ were added to the solution operating under SPEF conditions, the H2O2 concentration decreased strongly up to near 2 mM at 50 mA cm−2 (curve d). This strong loss in H2O2 can be related to its destruction from Fenton's reaction (5), also induced by photo-Fenton reaction (23), generating large amounts of •OH that effectively mineralize mecoprop, as will be discussed below. All these findings indicate that H2O2 can be produced at high enough rate in a BDD/GDE cell to remove relatively concentrated solutions of organic contaminants.
Mineralization and Energetic Parameters
The mineralization of organic pollutants in EAOPs is usually monitored from the abatement of its chemical oxygen demand (COD) or its total organic carbon (TOC). When H2O2 is generated, the measurement of TOC is preferable to avoid the analytical interference of this compound in COD. From these data, the percentage of TOC removal is calculated as follows [3,8]:
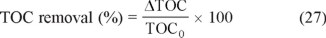
where ΔTOC is the experimental TOC decay (in mg L−1) at electrolysis time t and TOC0 is the starting value. The mineralization current efficiency (MCE) for an electrolyzed solution at a given time t (in h) is then estimated from Eq. (28) [34, 35]:
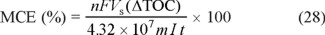
where n is the number of electrons exchanged during the mineralization of the checked organic, F is the Faraday constant (96485 C mol−1), Vs is the solution volume (in L), 4.32×107 is a conversion factor (= 3600 s h−1 × 12000 mg C mol−1), m is the number of carbon atoms of the molecule under study and I is the applied current (in A).
The viability of the EAOPs for industrial application is assessed from several energetic parameters. Operating at constant I, essential figures-of-merit are the energy consumption per unit volume (EC) and per unit TOC mass (ECTOC), which are calculated from Eqs. (29) and (30), respectively [36, 37]:
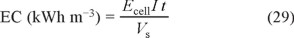
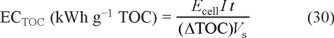
where Ecell is the average potential difference of the cell (in V).
Besides, when colored waters containing a dye are treated, the decay in color is usually determined from the decolorization efficiency or percentage of color removal by Eq. (31) [3]:
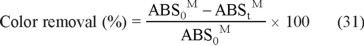
where ABS0M and ABStM denote the average absorbances before electrolysis and after an electrolysis time t, respectively, at the maximum visible wavelength (λmax) of the water.
EF Treatment with a BDD Anode of Organic Pollutants
Our pioneer work on EF with a BDD anode reported the superiority of this method compared to AO-H2O2 for the degradation of chlorophenoxy herbicides with a carbon-PTFE O2-fed (GDE) cathode [38]. Other authors have further considered both treatments using different cathodes [18, 39-44]. Several papers have also described the performance of EF alone for the degradation of some recalcitrant compounds [25, 28, 45-54]. Comparison of the oxidation ability of EF, PEF and SPEF will be discussed below. Figs. 2a and 2b depict the schemes of two typical stirred tank reactors used for such treatments with a BDD anode and H2O2 electrogeneration [55, 56].
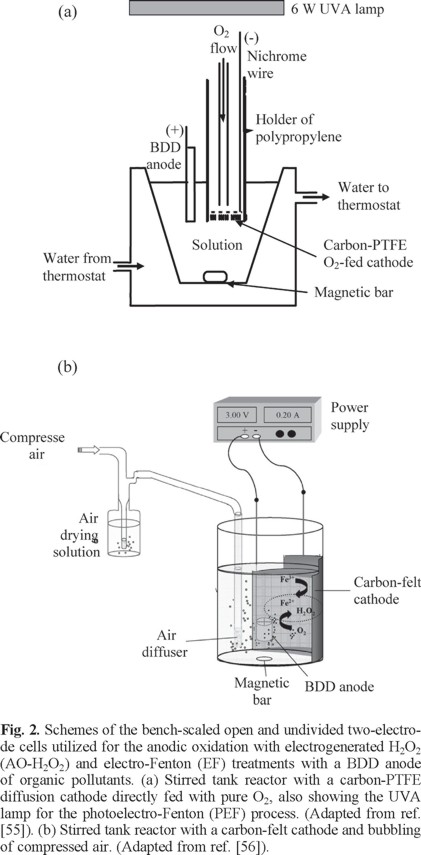
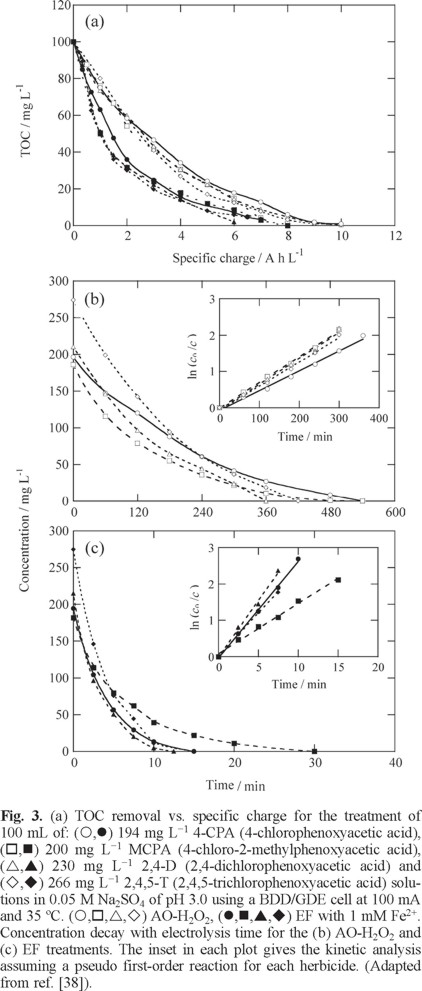
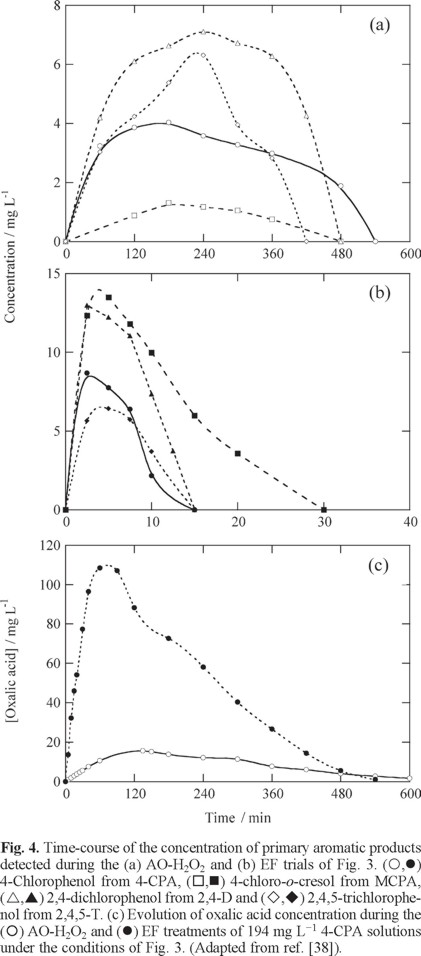
The large effectiveness of EF with a BDD anode and a GDE cathode was initially demonstrated using a stirred tank reactor like of Fig. 2a equipped with electrodes of 3 cm2. To do this, solutions containing 100 mg L−1 TOC of the herbicides 4-CPA (4-chlorophenoxyacetic acid), MCPA (4-chloro-2-methylphenoxyacetic acid), 2,4-D (2,4-dichlorophenoxyacetic acid) and 2,4,5-T (2,4,5-trichlorophenoxyacetic acid) in 0.05 M Na2SO4 of pH 3.0 and 35 ºC were comparatively treated by AO-H2O2 and EF with 1 mM Fe2+ by applying a current of 100 mA [38]. Fig. 3a highlights that the TOC of all solutions was removed at similar rate in each EAOP, attaining total mineralization after 6-8 h (6-8 A h L−1) of EF, a time slightly shorter than 9-10 h (9-10 A h L−1) needed for AO-H2O2. Although the degradation at the first stages (up to approximately 2 h) of EF was very fast, it needed practically the same time as AO-H2O2 to yield total mineralization. This can be related to the formation of complexes of Fe(III) with generated carboxylic acids in EF that are hardly oxidizable with BDD(•OH) and ●OH generated from reactions (18) and (5), respectively. Fig. 3b evidences a very slow decay of all herbicides in AO-H2O2 up to overall disappearance between 6 h for 2,4-D and 9 h for 2,4,5-T, a time slightly shorter than 9-10 h needed for their total TOC removal (see Fig. 3a), thereby suggesting the destruction of most oxidation products by BDD(●OH) at the same rate as generated. In contrast, Fig. 3c shows that all the herbicides were rapidly degraded by EF, disappearing between 12 min for 2,4-D and 30 min for MCPA. The quickest decay of herbicides in EF is then due to their reaction with greater amounts of •OH produced from Fenton's reaction (5). The kinetic analysis of the above results given in the insets of Figs. 3b and 3c indicate that all herbicides followed a pseudo-first-order reaction, with an apparent rate constant (k1) of 5.4x10−3- 7.1x10−3 min−1 for AO-H2O2 and as high as 0.12-0.31 min−1 for EF.
Reversed-phase HPLC allowed the detection of the primary phenolic byproduct of each herbicide, that is, 4-chlorophenol for 4-CPA, 4-chloro-o-cresol for MCPA, 2,4-dichlorophenol for 2,4-D and 2,4,5-trichlorophenol for 2,4,5-T. Figs. 4a and 4b show that the above phenols were continuously accumulated and destroyed at similar times to those required for the removal of the initial herbicides in AO-H2O2 and EF, respectively. In both methods phenols were then present in the medium while herbicides are destroyed. A very different behavior was found for oxalic acid, detected as the ultimate carboxylic acid by ion-exclusion chromatography. Fig. 4c exemplifies the evolution of this acid in the case of 4-CPA degradation. As can be seen, its concentration in AO-H2O2 only rose to 16 mg L−1 and decayed slowly up to its disappearance at 10 h, just when the solution TOC (see Fig. 3a) was completely removed. In EF this acid was accumulated in much larger extent because of the rapid degradation of aromatics and precedent carboxylic acids, then being slowly transformed into CO2 in 8 h, i.e., the same time as required for total 4-CPA mineralization (see Fig. 3a). This was ascribed to the fact that Fe(III)-oxalato complexes can only be attacked by BDD(•OH) since they are stable in EF with a Pt anode. The very slow destruction of such complexes on BDD then explains the difficulty of achieving total decontamination of herbicides by EF.
The superiority of EF over AO-H2O2 with a BDD anode has also been reported by other authors. Thus, Rodríguez De León et al. [40] used a cylindrical cell equipped with a 2 cm2 BDD anode and a 4 cm2 BDD cathode and filled with 40 mL of an air-saturated solution in 0.05 M Na2SO4 of pH 3.0 to comparatively decolorize and degrade 60 mg L−1 of the dyes Acid Yellow 36 and Methyl Orange. The decolorization processes always followed a pseudo-first-order reaction with an apparent rate constant (kapp) that increased largely in EF compared to AO-H2O2 when up to 0.3 mM Fe2+ was added to the solution, as a consequence of the additional oxidation with •OH formed in the bulk from Fenton's reaction (5). Abdessalem et al. [41] corroborated again the enhancement of organic degradation when •OH at the BDD surface and in the medium are simultaneously produced during the EF process with Fe3+ as catalyst. Air-saturated solutions of 250 mL with 0.125 mM of each of three pesticides chlortoluron, carbofuran and bentazone in 0.05 M Na2SO4 of pH 3.0 were treated in a stirred tank reactor like of Fig. 2b equipped with a 14 cm2 BDD anode and a 60 cm2 CF cathode. Under these conditions and operating at 300 mA, the three pesticides disappeared completely in 90-150 min in AO-H2O2, but only in 70-90 min in EF, always obeying a pseudo-first-order kinetics as detected by reversed-phase HPLC. Similarly, 90% TOC removal was achieved in 240 min of AO-H2O2, which was substantially reduced to 120 min in EF. At longer time, however, the reactivity of the remaining organic matter, mainly carboxylic acids, with hydroxyl radicals was so strongly inhibited that TOC was only reduced by 97-98% after 4 h of both treatments. Ion chromatography of degraded solutions revealed the total conversion of the initial Cl, N and S of the pesticides into Cl−, NH4+ and SO42− ions, respectively, although Cl− ion was slowly oxidized to Cl2.
The large effectiveness of the BDD anode has also been described for the destruction of the pesticide atrazine (2-chloro-4-ethylamino-6-isopropylamino-1,3,5-triazine), since this compound can only be transformed into cyanuric acid (2,4,6-trihydroxy-1,3,5-triazine) by chemical AOPs with in situ •OH production in the bulk. The study performed by Oturan et al. [43] demonstrated that using a stirred tank reactor like of Fig. 2b with a 25 cm2 BDD anode and applying a constant current of 1 A, the TOC of a 0.1 mM atrazine solution in 0.1 M Na2SO4 was reduced by 93% at pH 6.9 using AO-H2O2 and by 97% at pH 3.0 using EF with 0.1 mM Fe3+ as catalyst. In contrast, when a Pt anode was utilized in the EF process, only 83% of TOC removal was found under similar conditions. These results evidence that almost all the byproducts of atrazine, including cyanuric acid, can be destroyed by BDD(•OH) but not by Pt(•OH) formed from reaction (18), while the generation of •OH from Fenton's reaction (5) accelerates slightly the mineralization process.
It is also noteworthy the work of Montanaro et al. [39], who proposed the combination of the AO and EF processes to reduce the phosphorus content of an effluent from the manufacture of phosphorus-based flame retardants. A divided BDD/GDE cell with an anionic membrane was used. The method consisted in the AO treatment with a BDD anode of 100 mL of a fresh solution in the anolyte of the cell, followed by EF oxidation of this pretreated solution in the catholyte by •OH produced from Fenton's reaction (5). The anionic membrane allowed the passage of OH− ions from the cathodic to the anodic compartment to maintain the catholyte pH close to 1.5, thereby avoiding iron precipitation. A sequential running saved charge and time by using both anode and cathode performances in parallel, only needing 240 min at 10 mA cm−2 to decrease the phosphorus content below the limits needed.
The Peralta-Hernández's group has optimized the decolorization process of the azo dye Acid Yellow 36 by EF in a stirred tank reactor with a 2 cm2 BDD anode and cathode by means of response surface methodology (RSM) [25, 48]. Solutions of 100 mL containing 60, 70 and 80 mg L−1 of the dye were comparatively degraded in 0.05 M Na2SO4 as background electrolyte at initial pH 3.0 by applying a constant current density of 8, 15 and 23 mA cm−2 after addition of 0.1, 0.2 and 0.3 mM Fe2+. With these results, RSM was able to predict the optimal operating conditions to achieve 96% color removal with complete dye removal, which were 80 mg L−1 of the dye, 15 mA cm−2, 0.3 mM Fe2+ and 50 min of electrolysis time. This was confirmed with UV-vis and HPLC assessments during the EF treatment with BDD. More recently, this group has scaled-up the EF process to a recirculation pre-pilot plant of 3 L, similar to that of Fig. 1a but without photoreactor [53]. The BDD/BDD reactor contained electrodes of 64 cm2 area and solutions of 100, 150 and 200 mg L−1 Methyl Orange with 0.05 M Na2SO4 and 0.3 mM Fe2+ of pH 3.0 were decolorized at 7.8, 15 and 31 mA cm−2. After 60 min of electrolysis, the higher color removal of 70% was reached for 200 mg L−1 of Methyl Orange at 31 mA cm−2 due to the more effective degradation by •OH formed at the BDD anode and from Fenton's reaction (5). Ascorbic, benzoic, citric, maleic and oxalic acids were identified as intermediates by HPLC.
In collaboration between our group and Oturan's group, the antimicrobials chlorophene, triclocarban and triclosan were comparatively degraded by four EF systems consisting of Pt/GDE, BDD/GDE, Pt/CF and BDD/CF cells with Fe3+ as the catalyst [28,45]. In the cells with the GDE cathode (like of Fig. 2a), the pollutant decay was enhanced at greater Fe3+ content because this promoted quicker Fe2+ regeneration at the cathode with larger •OH production from Fenton's reaction (5). In contrast, when the CF cathode was used (cell of Fig. 2b), Fe2+ ion was largely accumulated as a result of the quick Fe3+ cathodic reaction (16) and so, only 0.2 mM Fe3+ was required to obtain the maximum •OH generation rate. Under these conditions, absolute rate constants of 1.00×1010 and 5.49×109 M−1 s−1 were obtained for the decay of chlorophene and triclosan, respectively. A poor mineralization degree was found for the Pt/GDE cell because of the difficult oxidation of final Fe(III)-oxalate complexes with •OH. These species were completely destroyed using the BDD/GDE cell at high current thanks to the great amount of reactive BDD(•OH) generated. Total mineralization was also achieved in both CF cells owing to the efficient oxidation of Fe(II)-oxalate complexes with •OH in the bulk. The higher oxidation ability was attained for the BDD/CF cell by the extra destruction of Fe(II)-oxalate complexes with BDD(•OH). Primary oxidation products like 2,4-dichlorophenol, 4-chlorocatechol, chlorohydroquinone and chloro-p-benzoquinone were identified for triclosan, whereas urea, hydroquinone, chlorohydroquinone, 1-chloro-4-nitrobenzene and 1,2-dichloro-4-nitrobenzene were found for triclocarban.
Our group has later considered the EF treatment of chlorinated aliphatic hydrocarbons such as 1,2-dichloroethane (DCA) and 1,1,2,2-tetrachloroethane (TCA) using a stirred tank reactor like of Fig. 2a [49]. Bulk electrolyses of 130 mL of 4 mM of both compounds in 0.035 M Na2SO4 with 0.5 mM Fe2+ at pH 3.0, 300 mA and 10 ºC yielded almost total mineralization in 420 min when a BDD/GDE cell was tested. Poorer degradation was obtained in comparative AO-H2O2 process or using a Pt anode in EF. Chloroacetic and dichloroacetic acids were the major by-products formed during the degradation of DCA and TCA, respectively. Acetic, oxalic and formic acids were also produced. Chlorine was released as Cl− ion, being further oxidized to ClO3− ion and, mostly, to ClO4− ion due to the action of the largely generated BDD(•OH) and •OH. Another study was carried out over the comparative degradation of the monoazo Acid Orange 7, diazo Acid Red 151 and triazo Direct Blue 71 by EF with a BDD/GDE cell [50]. The initial decolorization rate decreased with increasing initial azo bonds concentration due to the oxidation of more organic matter with similar amounts of hydroxyl radicals. Interestingly, this parameter lowered as the number of azo bonds in the molecule increased owing to their smaller reactivity with hydroxyl radicals. Reversed-phase HPLC revealed the total removal of all azo dyes following a pseudo first-order kinetics with rate constants showing the same trends as those predicted by initial decolorization rates. However, the decolorization process was slower as a consequence of the parallel destruction of colored conjugated intermediates formed during the EF treatment.
On the other hand, the Oturan's group has reported more fundamental information on the performance of EF with BDD using a cell like of Fig. 2b for the effective destruction of several organic pollutants like the dye Acid Orange 7 [46], the herbicide propham [47] and the antibiotic sulfachloropyridazine [51] in aqueous solutions, as well as the analgesic/anti-inflammatory ibuprofen in acetonitrile/water medium [54]. In these works, it is noticeable the determination of the absolute rate constant for the reaction of •OH with Acid Orange 7 (k2 = 1.1×1010 M−1 s−1) and sulfachloropyridazine (k2 = 1.6×109 M−1 s−1) using the competition kinetic method. Besides, intermediates and released inorganic ions were detected by GC-MS and HPLC. This allowed for each contaminant the proposal of a degradative route to explain its mineralization under the action of BDD(•OH) and •OH generated from reaction (18) and Fenton's reaction (5), respectively.
PEF Degradation with a BDD Anode of Organic Contaminants
The former work of our group on the PEF process with a BDD anode was focused to show its performance to destroy a toxic aromatic dye as Indigo Carmine (C.I. Acid Blue 64). The characteristics of the EF and PEF treatments of 100 mL of aqueous solutions containing 220 mg L−1 of this dye and Fe2+ and/or Cu2+ as catalysts at pH 3.0 with Pt/GDE and BDD/GDE cells like of Fig. 2a, with electrodes of 3 cm2 area, at 100 mA and 35.0 ºC were then studied [57]. While EF with Pt and 1.0 mM Fe2+ gave poor mineralization, only attaining 49% TOC removal in 540 min, EF with BDD promoted 91% TOC reduction at the same time, reaching total mineralization with loss of NH4+ ion in 780 min. This evidences again the much higher oxidation ability of BDD(•OH) compared with Pt(•OH) to remove organic matter. Indigo Carmine obeyed a pseudo-zero-order decay kinetics, as determined by HPLC, and disappeared at the same time as its aromatic derivatives isatin 5-sulphonic acid, indigo and isatin, mainly by reaction with •OH produced from Fenton's reaction (5). The most persistent byproducts were oxalic and oxamic acids, present in the medium as Fe(III)-oxalate and Fe(III)-oxamate complexes. Figs. 5a and b evidence that both complexes were poorly attacked with •OH and Pt(•OH) in EF with Pt, but they were completely destroyed with BDD(•OH) in EF with BDD, in agreement with the greater oxidation power of the latter radical. In PEF with Pt under a 6 W UVA light, TOC was reduced by 84% in 540 min and Fe(III)-oxalate complexes disappeared in 480 min (see Fig. 5a), but Fe(III)-oxamate complexes attained a steady concentration (see Fig. 5b). Consequently, the superiority of PEF compared to EF was related to the quick photolysis of Fe(III)-oxalate complexes from reaction (25). Cu2+ was then added as co-catalyst trying to produce additional quantity of •OH. It has been proposed that Cu2+ can be reduced to Cu+ with HO2• from reaction (32) and then, Cu2+ can be regenerated either from the Fenton-like reaction (33) yielding more •OH or from reaction (34) producing more Fe2+ to enhance Fenton's reaction (5) [58, 59]:
Cu2+ + HO2• → Cu+ + O2 + H+ (32)
Cu+ + H2O2 → Cu2+ + •OH + OH− (33)
Cu+ + Fe3+ → Cu2+ + Fe2+ (34)
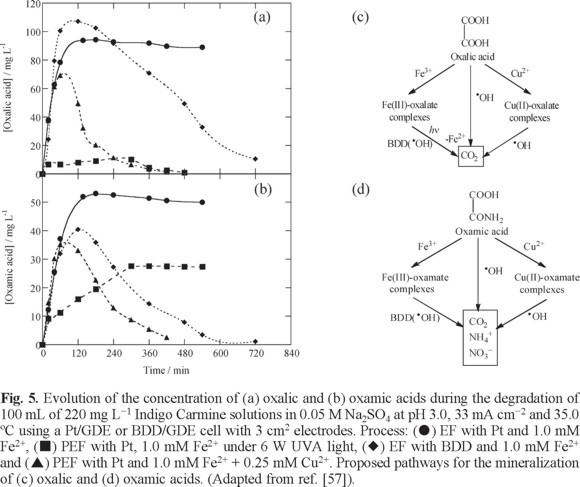
Surprisingly, PEF with Pt and 1.0 mM Fe2+ + 0.25 mM Cu2+ promoted almost total mineralization (> 97% TOC removal) in 450 min due to a faster and total destruction of both oxalic and oxamic acids (see Figs. 5a and b). The synergistic effect of Fe2+ and Cu2+ was explained by: (i) the photolysis of Fe(III)-oxalate complexes and (ii) the simultaneous reaction of competitively formed Cu(II)-oxalate and Cu(II)-oxamate complexes with •OH in the bulk. The proposed reaction pathways for the processes involved in the mineralization of oxalic and oxamic acids in the presence of both catalysts are shown in Figs. 5c and d, respectively.
The action of UVA light in the PEF process was further clarified by treating 100 mL of a 200 mg L−1 Direct Yellow 4 solution in 0.05 M Na2SO4 with 0.5 mM Fe2+ at pH 3.0, 33.3 mA cm−2 and 35 ºC in a similar BDD/GDE cell [60]. Fig. 6a depicts that the dye was not photodecomposed under a 6 W UVA irradiation, while the solution TOC was reduced by 84% and 97% after 360 min of EF and PEF, respectively. When the EF process was made for 60 min, 31% TOC was removed, but the Fenton's reagent remaining in the solution had oxidation ability enough to reduce the TOC by 45% up to 180 min, thus demonstrating the oxidation role of •OH formed from Fenton's reaction (5) in EF. In contrast, if the solution treated during 60 min in PEF was subsequently submitted to UVA irradiation, TOC abatement gradually increased up to 81%, whereas if the UVA light was applied after 120 min of EF, 97% TOC decay, equal to obtained in PEF, was found (see Fig. 6b). This is a very interesting finding, because it clearly shows that the photolytic action of UVA light, also involving the photodegradation of Fe(III)-oxalate complexes, acts pre-eminently at long electrolysis time of the PEF method. On the other hand, Almeida et al. [61] showed a quicker and total decolorization by PEF compared to EF for 100 mL of a 244 mg L−1 Acid Red 29 solution in 0.05 M Na2SO4 under optimum conditions of 0.5 mM Fe2+ and pH 3.0 at 35 ºC also using a stirred BDD/GDE tank reactor with 3 cm2 electrode area. The dye followed a pseudo-first-order decay with similar rate for EF and PEF. Since it disappeared much more rapidly than solution color, it was proposed that the decolorization process involved the destruction of colored conjugated products with λmax similar to that of Acid Red 29, which was enhanced in PEF by the production of more •OH from photo-Fenton reaction (23). Almost total mineralization was found for PEF, whereas poorer degradation was achieved by EF. In the PEF process, Fe(III)-oxalate complexes were efficiently photolyzed, but tartronic and oxamic acids were the most persistent byproducts because of the larger stability of their Fe(III) complexes. The initial N of the azo dye yielded NO3− ion, along with a smaller proportion of NH4+ ion.
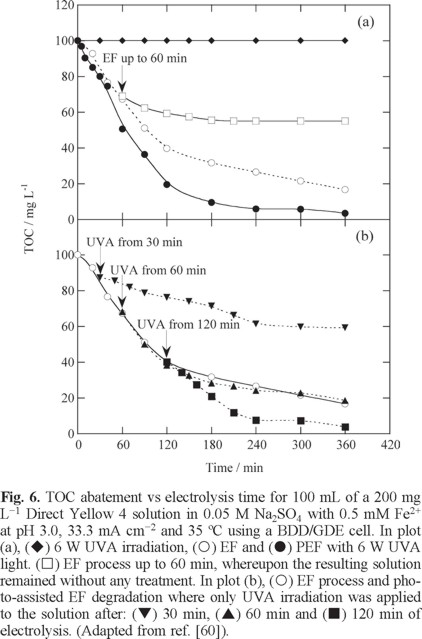
The great oxidation ability of PEF with BDD was also confirmed from the degradation of other chlorophenoxy herbicide such as 2,4-dichlorophenoxypropionic acid (2,4-DP) in a BDD/GDE cell [21]. The EF and PEF processes with 1 mM Fe2+ led to complete mineralization of 100 mL of 217 mg L−1 2,4-DP solutions at pH 3.0 300 mA and 35 ºC, with overall loss of chloride ion. Nevertheless, the UVA light in PEF had little effect on the degradation rate of pollutants compared with the fast oxidation produced by BDD(•OH) and •OH. As expected, the comparative procedures with Pt promoted slower decontamination because of the lower oxidizing power of this anode.
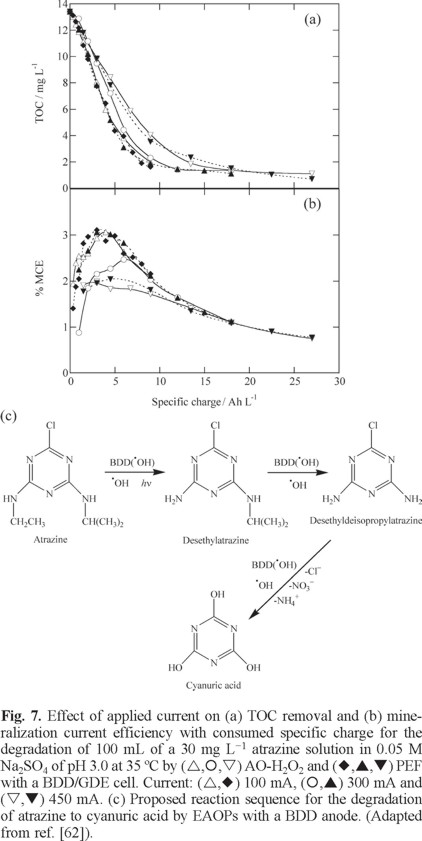
A further research of our group was devoted to the degradation of several s-triazinic herbicides such as atrazine [62], desmetryne (2-isopropylamino-4-methylamino-6-methylthio-1,3,5-triazine) [63] and cyanacine (2-(4-chloro-6-ethylamino-1,3,5-triazin-2-ylamino)-2-methylpropiononitrile) [64], which are difficultly oxidizable by AOPs because, as explained above, they are transformed into cyanuric acid that cannot be destroyed by •OH. The treatment of the three s-triazines with a stirred BDD/GDE tank reactor demonstrated that an almost total mineralization was always achieved by AO-H2O2, EF and PEF since cyanuric acid can be removed by generated BDD(•OH). This behavior can be observed in Fig. 7a for 30 mg L−1 atrazine, where 90-92% mineralization was attained in AO-H2O2 and PEF after 540 min of electrolysis at 100 mA and 360 min at 300 and 450 mA. The increase in current caused the spent of more specific charge, also reflected in the gradual fall in MCE, shown in Fig. 7b, suggesting a higher increase in rate of waste reactions (19)-(22). The low increase in TOC removal for PEF compared to AO-H2O2 was explained considering that the limiting oxidation reaction involved the attack of BDD(•OH) over cyanuric acid. For desmetryne and cyanazine, the effect of UVA light was more apparent because higher amounts of carboxylic acids like formic, oxamic and oxalic were produced from the degradation of their lateral groups. The optimum pH for all the processes was 2.0-4.0. Aromatic intermediates like desethylatrazine and desethyldesisopropylatrazine for atrazine, ammeline for desmetryne, and deisopropylatrazine, desethyldeisopropylatrazine and ammeline for cyanazine, which further evolved to cyanuric acid, were detected by HPLC. Fig. 7c exemplifies the reaction sequence proposed for the formation of this recalcitrant byproduct from atrazine [62]. In all cases, the initial N was mainly released as NO3− ion and the initial chlorine was lost as Cl− ion, which was oxidized to Cl2 at the BDD anode.
The effectiveness of the PEF process has been checked for an industrial chemical like sulfanilic acid (4-aminobenzenesulfonic acid) [65], which is widely used to synthesize pesticides, sulfonamide pharmaceuticals, sulfonated azo dyes, dye mordants, species, and food pigments. Using a stirred tank reactor like of Fig. 2a with a 3 cm2 BDD anode, it was found that 100 mL of 1.39 mM sulfanilic acid only underwent a partial decontamination of 85% by EF until 100 mA cm−2 at optimum conditions of 0.4 mM Fe2+ and pH 3.0. The rise in current density and substrate content led to an almost total mineralization. In contrast, the PEF process was more powerful giving almost total mineralization in less electrolysis time under comparable conditions. The kinetics for sulfanilic acid decay always obeyed a pseudo-first-order reaction. Hydroquinone and p-benzoquinone were detected as aromatic intermediates. Acetic, maleic, formic, oxalic and oxamic acids were identified and quantified as generated carboxylic acids. The fast photolysis of Fe(III)-carboxylate complexes, especially of the ultimate Fe(III)-oxalate and Fe(III)-oxamate species, under UVA radiation explained the greater oxidation ability of the PEF method. NH4+ ion was released in both treatments, along with NO3− ion in much smaller proportion.
The degradation of several emerging aromatic drugs including analgesics/anti-inflammatories such as ibuprofen [66] and salicylic acid [67], the blood lipid regulator metabolite clofibric acid [68], antimicrobials such as chloramphenicol [69], chloroxylenol [70], enrofloxacin [71], flumequine [72], sulfamethazine [24] and sulfanilamide [73], and β-blockers such as atenolol [74], propranolol [75, 76] and metoprolol [23], has also been tested. Table 1 summarizes the time needed for the total disappearance of pharmaceuticals (tTD) and the percentage of TOC removal at the end of comparative EF and PEF treatments of selected drug solutions in 0.05 M Na2SO4 at pH 3.0. Trials were carried out with stirred tank reactors like of Fig. 2a containing a Pt or BDD anode and a GDE cathode, with a 6 W UVA lamp for PEF. Two pairs of electrodes, also using a CF cathode to enhance the Fe2+ regeneration in the system from Fe3+ reduction by reaction (16), were checked for the β-blockers [74-76]. Inspection of Table 1 confirms the higher oxidation ability of the PEF method compared to EF for all the pharmaceuticals tested, since > 95% TOC abatement (related to almost total mineralization) was always found for the former process under comparable conditions. More mineralization degree was reached with a BDD anode instead of Pt, as expected for the higher oxidation power of generated BDD(•OH). The drugs decay verified pseudo-first-order reactions and in most cases, Table 1 evidences practically the same tTD values for both EF and PEF degradations, regardless of the anode (Pt or BDD) used. This indicates the pre-eminent reaction of aromatics with •OH formed from Fenton's reaction (5), but with poor production of this radical from photo-Fenton reaction (23). The greater TOC removal achieved in PEF is then due to the synergistic action of UVA light that is able to photolyze intermediates like Fe(III)-carboxylate complexes. Optimum degradation processes were obtained with 0.5-1.0 mM Fe2+ and at pH 3.0. It was also found that increasing current led to quicker mineralization due to the generation of more oxidizing species (Pt(•OH) or BDD(•OH) and •OH), but with the concomitant loss of MCE due to the higher rates attained for the waste reactions (19)-(22). The presence of more organic matter enhanced the current efficiency, although longer time was needed for a given TOC abatement.
Note the study performed for the β-blockers using undivided combined four-electrode cells containing a GDE and a CF cathode to enhance the mineralization process from the fast Fe2+ regeneration at the latter cathode [74-76]. Shorter disappearance times and higher percentages of TOC removal for atenolol and propranolol were attained for the combined Pt/GDE-Pt/CF and BDD/GDE-Pt/CF cells than for the single Pt/GDE and BDD/GDE ones in EF (see Table 1). Both drugs obeyed a pseudo-first-order kinetics with a rate at least 2.5 times higher in combined than in single cells. However, the fast destruction of Fe(III) complexes by UVA light in the analogous PEF processes led to similar mineralization degree for both kinds of cells when the most potent BDD anode was tested. This corroborates again the powerful synergistic influence of UVA irradiation in the latter method, which is even more potent to mineralize organic matter than the acceleration of Fenton's reaction (5) by increasing the rate of Fe2+ regeneration at the cathode. GC-MS and HPLC analysis of electrolyzed solutions allowed the detection of aromatic intermediates and generated carboxylic acids, leading to oxalic, oxamic and formic acids as ultimate byproducts. Fig. 8 presents the reaction sequence proposed for atenolol mineralization.
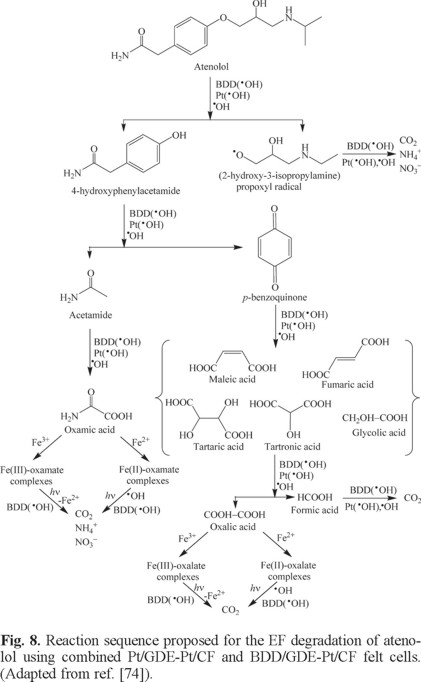
Degradation of Organic Pollutants by SPEF with a BDD anode
The SPEF process was proposed by our group to make a less expensive and more viable EAOP by using sunlight as renewable and inexpensive energy source. The removal of the drugs ibuprofen [66], salicylic acid [67], chloramphenicol [69] and enrofloxacin [71], as well as phthalic acid [77] which is an intermediate in the oxidation of naphthalenic rings, and the azo dye Sunset Yellow FCF [78] were comparatively studied by SPEF and other EAOPs (see Table 1) using a stirred tank reactor like of Fig. 2a equipped with a Pt or BDD anode and a GDE cathode, all with 3 cm2 area. In SPEF, the solution was submitted to an average UV intensity of sunlight of about 30-32 W m−2 and a mirror was placed at the bottom of the cell to better collect the solar energy.
Fig. 9a shows the TOC abatement with electrolysis time for the comparative EF, PEF and SPEF treatments of 100 mL of 41 mg L−1 ibuprofen with 0.5 mM Fe2+ at pH 3.0, 33.3 mA cm−2 and 25.0 ºC. As expected, each process was enhanced using a BDD anode instead of Pt due to the greater oxidizing power of BDD(•OH) than Pt(•OH) to remove the contaminants. Application of UV light promoted the degradation process and an almost total mineralization was reached more rapidly in SPEF by the higher intensity of UV radiation from sunlight. Similar results were found for the other organics tested, showing that SPEF with BDD always led to quicker degradation. For all the treatments, pH 3.0 was found optimal, near the optimum pH of 2.8 for Fenton's reaction (5), as expected if •OH is the main oxidant of organic pollutants. Higher amounts of TOC were removed with increasing current density and substrate concentration.
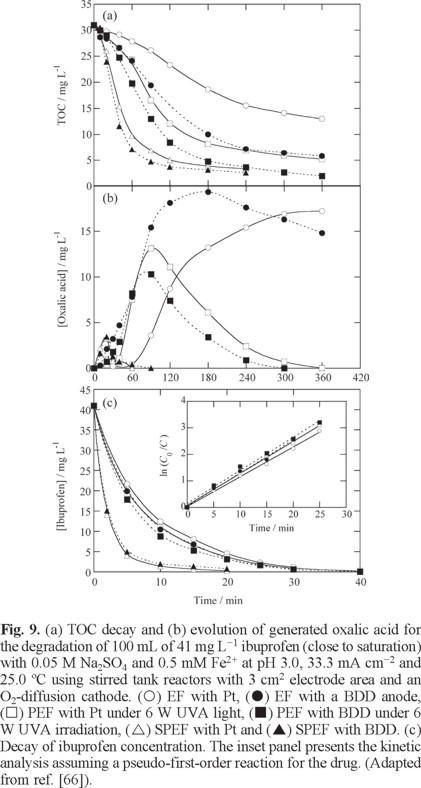
GC-MS, reversed-phase HPLC and/or LC-MS analysis of treated solutions revealed the formation of aromatic intermediates like 4-ethylbenzaldehyde, 4-isobutylacetophenone, 4-isobutylphenol and 1-(1-hydroxyethyl)-4-isobutylbenzene for ibuprofen; 2,3-, 2,5- and 2,6-dihydroxybenzoic acids for salicylic acid; 9 aromatic products and 13 hydroxylated derivatives for chloramphenicol; polyols, ketones and N-derivatives for enrofloxacin; 11 hydroxylated intermediates for phthalic acid; and 14 aromatic products and 34 hydroxylated derivatives, including benzenic, naphthalenic and phthalic acid compounds, for Sunset Yellow FCF. Ion-exclusion HPLC allowed the identification and quantification of different generated carboxylic acids, oxalic acid being the ultimate by-product accumulated in larger extent. Fig. 9b highlights the stability of the Fe(III)-oxalate complexes in EF, which were quickly removed in PEF and much more rapidly in SPEF by their fast photolysis from reaction (25). This behavior accounts for by the greatest mineralization degree attained in SPEF. Ion chromatography revealed that the initial F of enrofloxacin was completely transformed into F− ion and its initial N was primordially transformed into NH4+ ion and in smaller proportion into NO3− ion. The same fate was found for the initial N of chloramphenicol, but for Sunset Yellow FCF about 80% of initial N was loss from the solution, probably as N2 and NxOy species, suggesting a complex destruction of N-derivatives formed.
Fig. 9c illustrates a quite similar decay of ibuprofen concentration in EF and PEF, obeying a pseudo-first-order kinetics (see the inset panel), yielding k1 = 2.1×10−3 s−1. In contrast, the reaction is strongly enhanced in SPEF, as expected if much more •OH is produced from the action of the photo-Fenton reaction (23) as a result of the higher UV intensity of sunlight. This can also account for the higher oxidation ability of SPEF because primary products can be more rapidly destroyed leading intermediates that can be more quickly photolyzed by sunlight.
The SPEF treatment of organic pollutants was firstly scaled-up to the recirculation flow plant of 2.5 L with a BDD/GDE filter-press cell coupled to a flat solar photoreactor schematized in Fig. 1a [34-37, 71, 79, 80]. The electrodes with 20 cm2 area were separated 1.2 cm and the solar photoreactor was a polycarbonate box of 600 mL of irradiated volume, built-up with a mirror at the bottom and tilted 30° from the horizontal. Solutions with 50-300 mg L−1 of TOC in 0.05-0.10 M Na2SO4 with 0.5 mM Fe2+ at pH 3.0, 50-100 mA cm−2 and liquid flow rate of 180-200 L h−1 were usually tested. Table 2 summarizes the high TOC removals with excellent MCE values, along with the corresponding ECTOC values, determined for selected SPEF assays.
Similarly to found in the stirred tank reactor, SPEF was much more potent to mineralize organics than EF in the 2.5 L pre-pilot plant. As can be seen in Table 2, up to 637 mg L−1 of the pesticide mecoprop underwent 95% TOC reduction after a time as long as 540 min of SPEF at 50 mA cm−2 [35]. However, much faster degradation was found for the SPEF process of o-, m- and p-cresol, attaining 95-98% mineralization in only 180 min [34]. Analogously, almost total mineralization was also easily achieved for the dyes Acid Yellow 36 [36], Acid Red 88 [37], Acid Yellow 9 [37], Disperse Red 1 [79] and Disperse Red 3 [79] and for the pharmaceutical enrofloxacin [71]. For all these compounds, the MCE values increased at lower current density and higher contaminant concentration. For example, a maximum efficiency of 480% was obtained for the treatment of 1024 mg L−1 of o-cresol at 50 mA cm−2, which was reduced to 140% for 128 mg L−1. The very great MCE value found for this product evidences the great synergistic power of sunlight in SPEF. The same trend was obtained for ECTOC. While this parameter was as high as 0.259 kWh g−1 TOC after 240 min of EF degradation of 100 mg L−1 TOC of Disperse Red 1 at 50 mA cm−2, it was reduced to 0.151 kWh g−1 TOC (EC = 14.2 kWh m−3) for the comparative SPEF with 97% mineralization and 82% current efficiency. This corroborates that SPEF is much more economic than EF.
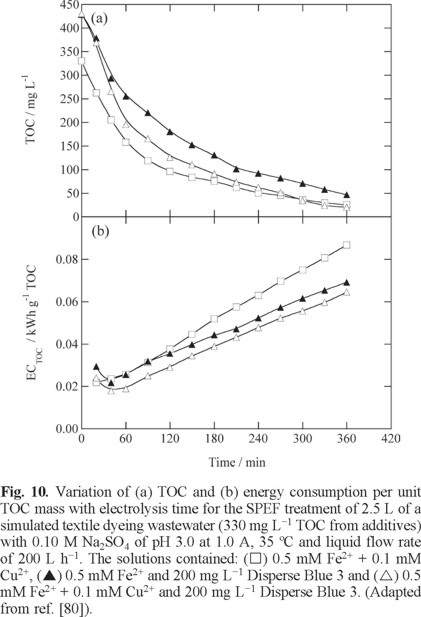
The performance of SPEF was enhanced by combining Fe2+ and Cu2+ as co-catalysts, as found for Disperse Blue 3 [80]. Figs. 10a and b illustrate that the use of 0.5 mM Fe2+ + 0.1 mM Cu2+. allowed > 95% TOC abatement in the presence and absence of 200 mg L−1 dye with ECTOC < 0.080 kWh g−1 TOC, more rapidly and less expensive than using 0.5 mM Fe2+ alone. This corroborates the attack of •OH on Cu(II) complexes of generated carboxylic acids like oxalic and oxamic (see Figs. 5c and d), competitively formed with Fe(III)-carboxylate ones.
All the organic pollutants tested in the 2.5 L pre-pilot plant dropped at similar rate in EF and SPEF obeying a pseudo-first-order kinetics and their k1 value increased with rising current density and decreasing pollutant concentrations. Aromatic intermediates were detected in several cases. For example, 2-methyl-p-benzoquinone for o- and m-cresol, 5-methyl-2-hydroxy-p-benzoquinone for p-cresol, 2-methylhydroquinone, 2-methyl-p-benzoquinone and 4-chloro-o-cresol for mecoprop and up to 15 anthraquinonic and phthalic acid derivatives for Disperse Blue 3. Analysis of final carboxylic acids confirmed the quick photolysis of Fe(III)-oxalato complexes, but the formation of other more recalcitrant acids like acetic and oxamic slowed the mineralization process. Cl− ion was loss in mecoprop degradation [35], whereas SO42−, NH4+ and/or NO3− ions were loss in the mineralization of dyes [36,37,78].
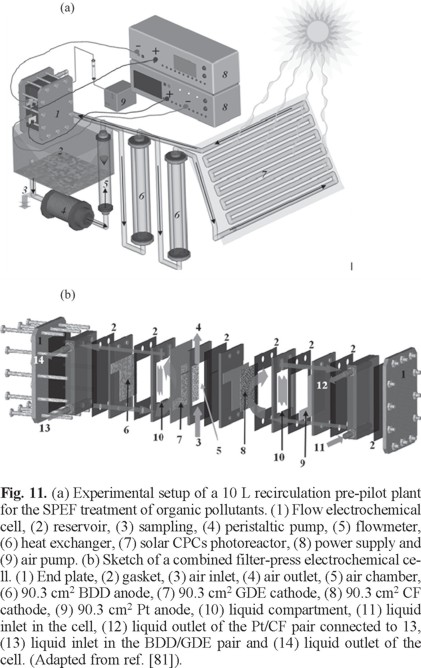
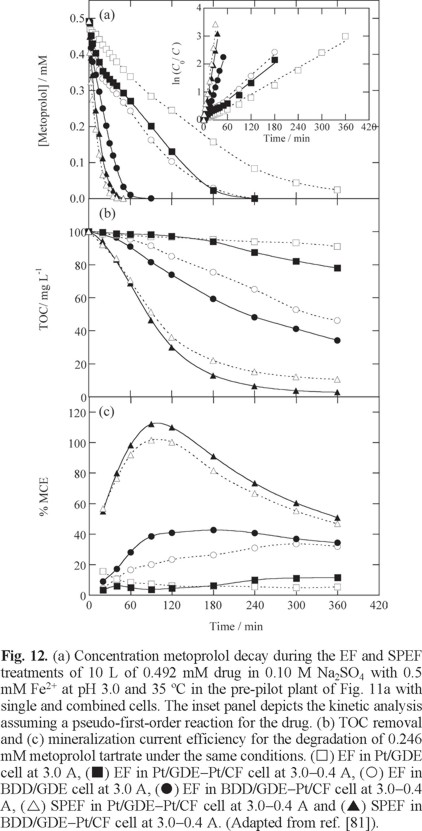
The study of the SPEF process was lately extended to a 10 L pre-pilot plant, schematized in Fig. 11a, where the electrochemical filter-press reactor contained electrodes of 90.3 cm2 area and was coupled to a 1.57 L compound parabolic collectors (CPCs) as the solar photoreactor. This plant has been used by Isarain et al. [81] to extent the study of the SPEF degradation of 100 mg L−1 TOC of solutions with the β-blockers atenolol, metoprolol tartrate and propranolol hydrochloride in 0.10 M Na2SO4 with 0.5 mM Fe2+ at pH 3.0 using single Pt/ADE and BDD/ADE cells and their combination with a Pt/CF cell to enhance Fe2+ regeneration from Fe3+ reduction. As an example, Fig. 11b shows a sketch of the combined BDD/GDE-Pt/CF cell used. For metoprolol tartrate, Figs. 12a, b and c evidence the superiority of combined cells over single ones, BDD over Pt and SPEF over EF regarding the decay kinetics of the substrate, TOC removal and MCE, respectively. This can be related to the larger generation of •OH from Fenton's reaction (5) in the combined cells, the higher oxidizing power of BDD(•OH) and the photolytic action of sunlight in SPEF, as can also be deduced from the relative pseudo-first-order decay kinetics shown in the inset of Fig. 12a for the different cells checked. Although the combined BDD/GDE-Pt/CF cell was the most potent system, the lowest ECTOC of 0.080 kWh g−1 TOC with 88-93% mineralization was obtained for the Pt/GDE-Pt/CF one.
Conclusions
EAOPs based on Fenton's reaction chemistry such as EF, PEF and SPEF are viable to remove toxic and refractory organic pollutants from acidic waters because they allow high mineralization rates with good current efficiency. These simple, safe and environmentally friendly technologies can be easily scaled-up from stirred tank reactors to industrial level using recirculation flow plants. The current electrochemical technology permits the use of very stable and powerful anodes like BDD thin films and several carbonaceous cathodes for an efficient H2O2 generation. The main drawback for industrial application of these EAOPs is the electrical cost for running the electrochemical cell since the energy cost required for the UV lamp in PEF can be avoided when sunlight is used in SPEF. The possible coupling of photovoltaic systems to provide inexpensive electrical energy to the electrochemical reactor could be an excellent alternative way that may be tested in the next future to solve this problem. The coupling of the reactor with efficient solar CPCs photoreactors is another interesting possibility for enhancing the degradation process. It has been shown that the SPEF treatment of organic pollutants with a BDD anode is more efficient and less expensive than using other EAOPs under comparable conditions. In EF, PEF and SPEF with BDD, the mineralization rate was slowed as applied current dropped and pollutant concentration increased, but with greater MCE and lower energy consumption. The contaminant decay usually followed a pseudo-first-order kinetics. Aromatic intermediates were oxidized by generated •OH giving short-linear carboxylic acids that form Fe(III) complexes. Most of Fe(III)-carboxylate species were much more rapidly photolyzed by the more potent UV radiation supplied by sunlight in SPEF than by artificial UVA lamps in PEF. Heteroatoms present in organics were also mineralized and released as inorganic ions such as F−, Cl−, SO42−, NH4+ and NO3−.
Acknowledgements
The author thanks financial support from MICINN (Ministerio de Ciencia e Innovación, Spain) under project CTQ2010-16164/ BQU, co-financed with FEDER funds.
References
1. Khetan, S. K.; Collins, T. J. Chem. Rev. 2007, 107, 2319. [ Links ]
2. Martínez-Huitle, C. A.; Brillas, E. Appl. Catal. B: Environ. 2009, 87, 105. [ Links ]
3. Kümmerer, K. Chemosphere 2009, 75, 417. [ Links ]
4. Luo, Y.; Xu, L.; Rysz, M.; Wang, Y.; Zhang, H.; Alvarez, P. J. J. Environ. Sci. Technol. 2011, 45, 1827. [ Links ]
5. Esplugas, S.; Bila, D. M.; Krause, L. G. T.; Dezotti, M. J. Hazard. Mater. 2007, 149, 631. [ Links ]
6. Klavarioti, M.; Mantzavinos, D.; Kassinos, D. Environ. Int. 2009, 35, 402. [ Links ]
7. Panizza, M.; Cerisola, G. Chem. Rev. 2009, 109, 6541. [ Links ]
8. Brillas, E.; Sirés, I.; Oturan, M. A. Chem. Rev. 2009, 109, 6570. [ Links ]
9. Serra, A.; Domènech, X.; Arias, C.; Brillas, E.; Peral, J. Appl. Catal. B: Environ. 2009, 89, 12. [ Links ]
10. Sirés, I.; Brillas, E. Environ. Int. 2012, 40, 212. [ Links ]
11. Pletcher, D. Acta Chem. Scand. 1999, 53, 745. [ Links ]
12. Foller, P. C.; Bombard, R. T. J. Appl. Electrochem. 1995, 25, 613. [ Links ]
13. Alvarez Gallegos, A.; Vergara García, Y.; Zamudio, A. Sol. Energy Mater. Sol. Cells 2005, 88, 157. [ Links ]
14. Khataee, A. R.; Zarei, M. Desalination 2011, 278, 117. [ Links ]
15. Khataee, A. R.; Vahid, B.; Behjati, B.; Safarpour, M. Environ. Progr. Sust. Energy 2013, 32, 557. [ Links ]
16. Khataee, A. R.; Safarpour, M.; Naseri, A.; Zarei, M. J. Electroanal. Chem. 2012, 672, 53. [ Links ]
17. Kaplan, F.; Hesenov, A.; Gozmen, B.; Erbatur, O.; Environ. Technol. 2011, 32, 685. [ Links ]
18. Murati, M.; Oturan, N.; Aaron, J. J.; Dirany, A.; Tassin, B.; Zdravkovski, Z.; Oturan, M. A. Environ. Sci. Pollut. Res. 2012, 19, 1563. [ Links ]
19. Wang, A.; Qu, J.; Liu, H.; Ru, J. Appl. Catal. B: Environ. 2008, 84, 393. [ Links ]
20. Wang, A.; Li, Y. -Y.; Estrada, A. L. Appl. Catal. B: Environ. 2011, 102, 378. [ Links ]
21. Brillas, E.; Baños, M. A.; Skoumal, M.; Cabot, P. L.; Garrido, J.A.; Rodríguez, R. M. Chemosphere 2007, 68, 199. [ Links ]
22. Expósito, E.; Sánchez-Sánchez, C. M.; Montiel, V. J. Electrochem. Soc. 2007, 154, E116. [ Links ]
23. Isarain-Chávez, E.; Garrido, J. A.; Rodríguez, R. M.; Centellas, F.; Arias, C.; Cabot, P. L.; Brillas, E. J. Phys. Chem. A 2011, 115, 1234. [ Links ]
24. El-Ghenymy, A.; Rodríguez, R. M.; Arias, C.; Centellas, F.; Garrido, J. A.; Cabot, P. L.; Brillas, E. J. Electroanal. Chem. 2013, 701, 7. [ Links ]
25. Cruz-González, K.; Torres-López, O.; García-León, A. M.; Brillas, E.; Hernández-Ramírez, A.; Peralta-Hernández, J. M. Desalination 2012, 286, 63. [ Links ]
26. Sun, Y.; Pignatello, J. J. Environ. Sci. Technol. 1993, 27, 304. [ Links ]
27. Oturan, M. A.; Oturan, N.; Aaron, J. J. Actual. Chimique 2004, 277-278, 57. [ Links ]
28. Sirés, I.; Garrido, J. A.; Rodríguez, R. M.; Brillas, E.; Oturan, N.; Oturan, M. A. Appl. Catal. B: Environ. 2007, 72, 382. [ Links ]
29. Marselli, B.; Garcia-Gomez, J.; Michaud, P. A.; Rodrigo, M. A.; Comninellis, Ch. J. Electrochem. Soc. 2003, 150, D79. [ Links ]
30. Panizza, M.; Cerisola, G. Electrochim. Acta, 2005, 51, 191. [ Links ]
31. Panizza, M.; Michaud, P. A.; Cerisola, G.; Comninellis, Ch. J. Electroanal. Chem. 2001, 507, 206. [ Links ]
32. Flox, C.; Cabot, P. L.; Centellas, F.; Garrido, J. A.; Rodríguez, R. M.; Arias, C.; Brillas, E. Chemosphere 2006, 64, 892-902. [ Links ]
33. Zuo, Y.; Hoigné, J. Environ. Sci. Technol. 1992, 26, 1014. [ Links ]
34. Flox, C.; Cabot, P. L.; Centellas, F.; Garrido, J. A.; Rodríguez, R. M.; Arias, C.; Brillas, E. Appl. Catal. B: Environ. 2007, 75, 17. [ Links ]
35. Flox, C.; Garrido, J. A.; Rodríguez, R. M.; Cabot, P. L.; Centellas, F.; Arias, C.; Brillas, E. Catal. Today 2007, 129, 29. [ Links ]
36. Ruiz, E. J.; Arias, C.; Brillas, E.; Hernández-Ramírez, A.; Peralta-Hernández, J. M. Chemosphere 2011, 82, 495. [ Links ]
37. Ruiz, E. J.; Hernández-Ramírez, A.; Peralta-Hernández, J. M.; Arias, C.; Brillas, E. Chem. Eng. J. 2011, 171, 385. [ Links ]
38. Brillas, E.; Boye, B.; Sirés, I.; Garrido, J. A.; Rodríguez, R. M.; Arias, C.; Cabot, P. -L.; Comninellis, Ch. Electrochim. Acta 2004, 49, 4487. [ Links ]
39. Montanaro, D.; Petrucci, E.; Merli, C. J. Appl. Electrochem. 2008, 38, 947. [ Links ]
40. Rodríguez De León, N. E.; Cruz-González, K.; Torres-López, O.; Ramírez-Hernández, A.; Guzmán-Mar, J. L.; Martínez-Huitle, C. A.; Peralta-Hernández, J. M. ECS Transactions 2009, 20, 283. [ Links ]
41. Abdessalem, A. K.; Oturan, M. A.; Oturan, N.; Bellakhal, N.; Dachraoui, M. Int. J. Environ. Anal. Chem. 2010, 90, 468. [ Links ]
42. Oturan, N.; Hamza, M.; Ammar, S.; Abdelhedi, R.; Oturan, M. A. J. Electroanal. Chem. 2011, 661, 66. [ Links ]
43. Oturan, N.; Brillas, E.; Oturan, M. A. Environ. Chem. Lett. 2012, 10, 165. [ Links ]
44. Almomani, F.; Baranova, E. A. Water Sci. Technol. 2012, 66, 465. [ Links ]
45. Sirés, I.; Oturan, N.; Oturan, M. A.; Rodríguez, R. M.; Garrido, J. A.; Brillas, E. Electrochim. Acta 2007, 52, 5493. [ Links ]
46. Hammami, S.; Bellakhal, N.; Oturan, N.; Oturan, M. A.; Dachraoui, M. Chemosphere 2008, 73, 678. [ Links ]
47. Ozcan, A.; Sahin, Y.; Koparal, A. S.; Oturan, M. A. Appl. Catal. B: Environ. 2009, 89, 620. [ Links ]
48. Cruz-González, K.; Torres-López, O.; García-León, A.; Guzmán-Mar, J. L.; Reyes, L. H.; Hernández-Ramírez, A.; Peralta-Hernández, J. M. Chem. Eng. J. 2010, 160, 199. [ Links ]
49. Randazzo, S.; Scialdone, O.; Brillas, E.; Sirés, I. J. Hazard. Mater. 2011, 192, 1555. [ Links ]
50. Garcia-Segura, S.; Centellas, F.; Arias, C.; Garrido, J. A.; Rodríguez, R. M.; Cabot, P. L.; Brillas, E. Electrochim. Acta 2011, 58, 303. [ Links ]
51. Dirany, A.; Sirés, I.; Oturan, N.; Ozcan, A.; Oturan, M. A. Environ. Sci. Technol. 2012, 46, 4074. [ Links ]
52. Almomani, F.; Baranova, E. A. Environ. Technol. 2013, 34, 1473. [ Links ]
53. Isarain-Chavez, E.; de la Rosa, C.; Martínez-Huitle, C. A.; Peralta-Hernández, J. M. Int. J. Electrochem. Sci. 2013, 8, 3084. [ Links ]
54. Loaiza-Ambuludi, S.; Panizza, M.; Oturan, N.; Ozcan, A.; Oturan, M. A. J. Electroanal. Chem. 2013, 702, 31. [ Links ]
55. Brillas, E.; Bastida, R. M.; Llosa, E.; Casado, J. J. Electrochem. Soc. 1995, 142, 1733. [ Links ]
56. Oturan, M. A.; Guivarch, E.; Oturan, N.; Sirés, I. Appl. Catal. B: Environ. 2008, 82, 244. [ Links ]
57. Flox, C.; Ammar, S.; Arias, C.; Brillas, E.; Vargas-Zavala, A. V.; Abdelhedi, R. Appl. Catal. B: Environ. 2006, 67, 93. [ Links ]
58. Sharma, V. K.; Millero, F. J. Environ. Sci. Technol. 1988, 22, 768. [ Links ]
59. Gallard, H.; De Laat, J.; Legube, B. Rev. Sci. Eau 1999, 12, 713. [ Links ]
60. Garcia-Segura, S.; El-Ghenymy, A.; Centellas, F.; Rodríguez, R. M.; Arias, C.; Garrido, J. A.; Cabot, P. L.; Brillas, E. J. Electroanal. Chem. 2012, 681, 36. [ Links ]
61. Almeida, L. C.; Garcia-Segura, S.; Arias, C.; Bocchi, N.; Brillas, E. Chemosphere 2012, 89, 751. [ Links ]
62. Borràs, N.; Oliver, R.; Arias, C.; Brillas, E. J. Phys. Chem. A 2010, 114, 6613. [ Links ]
63. Borras, N.; Arias, C.; Oliver, R.; Brillas, E. Chemosphere 2011, 85, 1167. [ Links ]
64. Borràs, N.; Arias, C.; Oliver, R.; Brillas, E. J. Electroanal. Chem. 2013, 689, 158. [ Links ]
65. El-Ghenymy, A.; Garrido, J. A.; Centellas, F.; Arias, C.; Cabot, P. L.; Rodríguez, R. M.; Brillas, E. J. Phys. Chem. A 2012, 116, 3404. [ Links ]
66. Skoumal, M.; Rodríguez, R. M.; Cabot, P. L.; Centellas, F.; Garrido, J. A.; Arias, C.; Brillas, E.; Electrochim. Acta 2009, 54, 2077. [ Links ]
67. Guinea, E.; Arias, C.; Cabot, P. L.; Garrido, J. A.; Rodríguez, R. M.; Centellas, F.; Brillas, E.; Water Res. 2008, 42, 499. [ Links ]
68. Sirés, I.; Centellas, F.; Garrido, J. A.; Rodríguez, R. M.; Arias, C.; Cabot, P. L.; Brillas, E. Appl. Catal. B: Environ. 2007, 72, 373. [ Links ]
69. Garcia-Segura, S.; Cavalcanti, E. B.; Brillas, E. Appl. Catal. B: Environ. 2014, 144, 588. [ Links ]
70. Skoumal, M.; Arias, C.; Cabot, P. L.; Centellas, F.; Garrido, J. A.; Rodríguez, R. M.; Brillas, E. Chemosphere 2008, 71, 1718. [ Links ]
71. Guinea, E.; Garrido, J. A.; Rodríguez, R. M.; Cabot, P. L.; Arias, C.; Centellas, F.; Brillas, E. Electrochim. Acta 2010, 55, 2101. [ Links ]
72. Garcia-Segura, S.; Garrido, J. A.; Rodríguez, R. M.; Cabot, P. L.; Centellas, F.; Arias, C.; Brillas, E. Water Res. 2012, 46, 2067. [ Links ]
73. El-Ghenymy, A.; Oturan, N.; Oturan, M. A.; Garrido, J. A.; Cabot, P. L.; Centellas, F.; Rodríguez, R. M.; Brillas, E. Chem. Eng. J. 2013, 234, 115. [ Links ]
74. Isarain-Chávez, E.; Arias, C.; Cabot, P. L.; Centellas, F.; Rodríguez, R. M.; Garrido, J. A.; Brillas, E. Appl. Catal. B: Environ. 2010, 96, 361. [ Links ]
75. Isarain-Chávez, E.; Rodríguez, R. M.; Garrido, J. A.; Arias, C.; Centellas, F.; Cabot, P. L.; Brillas, E. Electrochim. Acta 2010, 56, 215. [ Links ]
76. Isarain-Chávez, E.; Cabot, P. L.; Centellas, F.; Rodríguez, R. M.; Arias, C.; Garrido, J. A.; Brillas, E. J. Hazard. Mater. 2011, 185, 1228. [ Links ]
77. Garcia-Segura, S.; Salazar, R.; Brillas, E. Electrochim. Acta 2013, 113, 609. [ Links ]
78. Moreira, F. C.; Garcia-Segura, S.; Vilar, V. J. P.; Boaventura, R. A. R.; Brillas, E. Appl. Catal. B: Environ. 2013, 142-143, 877. [ Links ]
79. Salazar, R.; Garcia-Segura, S.; Ureta-Zañartu, M. S.; Brillas, E. Electrochim. Acta 2011, 56, 6371. [ Links ]
80. Salazar, R.; Brillas, E.; Sirés, I. Appl. Catal. B: Environ. 2012, 115-116, 107. [ Links ]
81. Isarain-Chávez, E.; Rodríguez, R. M.; Cabot, P. L.; Centellas, F.; Arias, C.; Garrido, J. A.; Brillas, E. Water Res. 2011, 45, 4119. [ Links ]

