










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkJournal of the Mexican Chemical Society
Print version ISSN 1870-249X
J. Mex. Chem. Soc vol.58 n.2 Ciudad de México Apr./Jun. 2014
Article
NMR and Theoretical Studies on the Conformational Preferences of Some Non-metal Coordinated N-Enoyl Systems Attached to Common Chiral Auxilaries
Rosmarbel Morales-Nava,1,3 Alejandro Ramírez-Solís,2+ and Mario Fernández-Zertuche1*
1 Centro de Investigaciones Químicas. Universidad Autónoma del Estado de Morelos. Av. Universidad 1001, Col. Chamilpa, Cuernavaca, Morelos, C.P 62209, México. mfz@uaem.mx
2 Laboratoire de Physico-chimie de Nano-objets, INSA-Université de Toulouse. 135 Av. de Rangueil, Toulouse 31065, France.
+ On sabbatical leave from Facultad de Ciencias. Universidad Autónoma del Estado de Morelos.
3 Instituto de Ciencias Físicas. Universidad Nacional Autónoma de México. Av. Universidad S/N, Col. Chamilpa, Cuernavaca, Morelos, C.P. 62210, México.
Received April 4th, 2013
Accepted November 11th, 2013
Abstract
We report a systematic study of a series of N-enoyl systems attached to common oxazolidin-2-ones, oxazolidine-2-thiones and thiazolidine-2-thiones chiral auxiliaries in order to determine the most stable conformation of these compounds. 1H NMR studies show an anti-s-cis structure as the most stable conformation for these series of compounds. Density Functional Theory geometry optimizations and vibrational analysis using the B3LYP exchange-correlation functional with the standard 6-31G** basis sets were done, including solvent effects (chloroform and toluene). Gibbs free energy differences show that the anti-s-cis structures are the most stable conformers lying, on average, ca. 6 kcal/mol lower in energy than the syn-s-cis conformers, widely used to explain the structure and reactivity of N-enoyl systems.
Key words: Enoyl systems, conformational preferences, chiral auxiliaries, NMR shifts.
Resumen
Se reporta un estudio sistemático sobre una serie de sistemas tipo N-enoilo, unidos a algunos auxiliares quirales comunes como las oxazolidin-2-onas, oxazolidin-2-tionas y tiazolidin-2-tionas para determinar la conformación más estable de estos compuestos. Estudios de RMN 1H muestran a la conformación anti-s-cis como la más estable para estas series de compuestos. En el marco de la Teoría de Funcionales de la Densidad se realizaron optimizaciones de geometría y análisis vibracional con el funcional B3LYP y las bases 6-31G**, incluyendo los efectos del disolvente en cloroformo y tolueno. Las diferencias de energía libre de Gibbs muestran que las conformaciones anti-s-cis son las más estables, en promedio, ~6 kcal/mol más estables que los confórmeros syn-s-cis, generalmente usados para explicar la estructura y reactividad de sistemas tipo N-enoilo.
Palabras clave: Sistemas enoilo, preferencias conformacionales, auxiliares quirales, desplazamientos RMN.
Introduction
The 1,4-addition of nucleophiles to α,β-unsaturated carboxylic acid derivatives is one of the most useful methods for asymmetric carbon-carbon or carbon-heteroatom bond formation [1]. In this context, the conjugate addition of organometallic reagents to N-enoyloxazolidinone systems has been employed in the synthesis of natural products [2]. Metal coordination with one or both oxygens on these systems enhances the electrophilic character of the β-carbon, lowering the energy of the molecular orbitals of the conjugated systems and locking these substrates in a specific conformation to ensure facial selectivity during the addition [3].
Nevertheless, in the conjugate addition of some organocopper reagents to this kind of substrates, Williams [4] and Bergdahl [5] have reported a reversed diastereofacial selectivity on some usual 4-alkyl substituted oxazolidinones of the same relative configurations. They suggest that the observed different stereochemical outcomes reflect the presence of different conformers of the unsaturated systems. (Fig. 1).
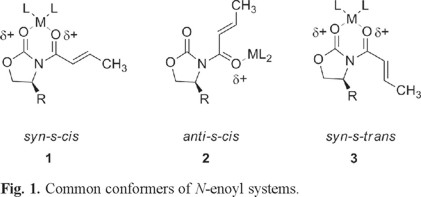
More recently, Sabala et al [6] suggests that N-enoyloxazolidinone systems in an anti-s-cis conformation lead to syn addition products, whereas a syn-s-cis conformation lead to anti addition products. In both cases addition of the nucleophile always takes place on the face of the double bond opposite to the R group in the chiral auxiliary. (Fig. 2)
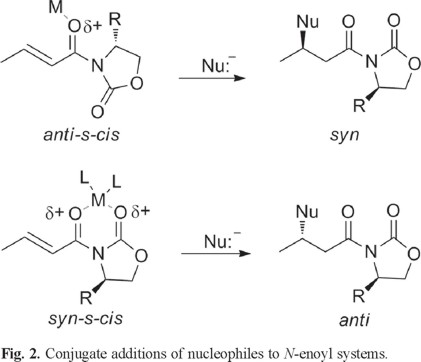
Moreover, when exploring the synthetic utility of N-enoyloxazolidinethiones, the addition of cuprates in the presence of TMSI yields anti addition products, suggesting a syn-s-cis conformation for this kind of substrates. In the case of N-enoylthiazolidinethiones [7], only one example of cuprate addition has appeared in the literature where the addition product seems to be derived from an anti-s-cis conformation of the reacting substrate.
Within the context of our medicinal chemistry program whose main goal is the design and synthesis of structural analogues of well known pharmacologically active agents, we became interested in this type of analogues, since they are versatile precursors to some active agents.
Although the conformation of the metal coordinated N-enoyl and similar species and their 1H NMR spectra have received some attention in the literature [8], to the best of our knowledge, the conformational preferences and energy differences between the possible conformers of the free non-metal coordinated N-enoyl systems 4,5,6 and 7 (Fig. 3) have not been documented.
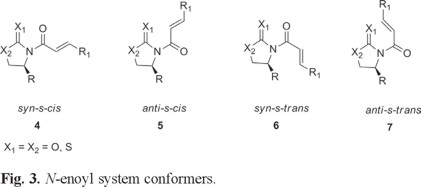
Intrigued by the different stereochemical behaviors reported in the literature on the addition of cuprates to these systems, we carried out some 1H NMR and theoretical calculations in order to determine the most stable conformers of these conjugated systems [4, 9d].
Results and Discussion
1H NMR Studies
Table 1 shows the compounds chosen for this study, 8-10. All of them have been prepared following standard literature procedures [2a, 9].
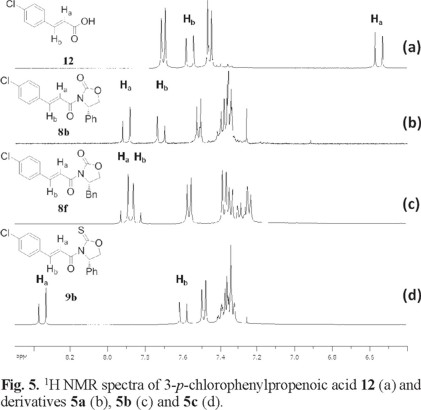
The 1H NMR spectra of α,β-unsaturated derivatives can provide some insight on their conformational preference based on the chemical shifts of the vinylic protons, Ha and Hb. Fig. 4 and 5 show the 1H NMR spectra of some illustrative examples of these compounds. Fig. 4 a and 5 a show the 1H NMR spectra of crotonic acid 11 and of 4-p-chlorophenylpropenoic acid 12 respectively, showing the chemical shift of Ha and Hb for these acids and their derivatives.
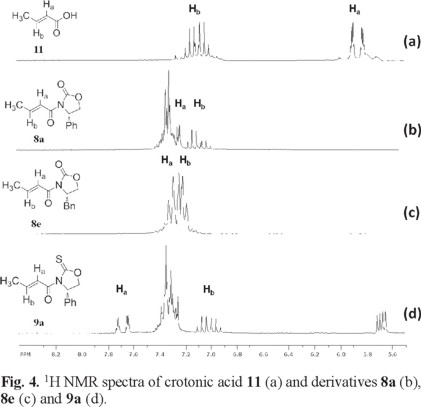
As can be seen in Fig 4, the chemical shift of Ha in crotonic acid appears at δ 5.85 ppm (Fig. 4 a). However, when the chiral auxiliaries are coupled to crotonic acid, the chemical shift of Ha suffers a significant high frequency shift in their 1H NMR spectra. For example, when (4S)-4-phenyl-1,3-oxazolidine-2-one, (4S)-4-benzyl-1,3-oxazolidine-2-one or (4S)-4-phenyl-1,3-oxazolidine-2-thione are coupled to acid 11, 1H NMR signal for Ha suffers the high frequency to δ 7.26 in compound 8a; to δ 7.30 ppm in compound 8e and to δ 7.68 in compound 9a (Fig 4 b-d). In compound 8e, Ha and Hb were assigned based on their coupling constants, for example, Ha exhibits a single coupling constant of 15.0 Hz showing its coupling to Hb. On the other hand, for Hb two coupling constants can be observed, one of them has a magnitude of 15.0 Hz as expected and the other one showing a coupling constant of 5.2 Hz due to its coupling with the methyl group protons. This is consistent with the 1H NMR spectra reported in the literature for this compound [10].
Fig 5 shows the 1H NMR spectra of 4-p-chlorophenylpropenoic acid 12 where the chemical shift of Ha appears at δ 6.54 ppm (Fig. 5 a). In a similar manner, when the same chiral auxiliaries are coupled to acid 11, compounds 8b, 8f and 9b are obtained. Once again the chemical shifts of Ha in these compounds suffer the high frequency shift to δ 7.90 in 8b, to δ 7.90 in 8f and to δ 8.35 in 9b (Fig. 5 b-d). In all these cases, this represents an average downfield displacement of δ 1.53.
On the other hand, although the chemical shift of Hb experiences some minor displacement, it practically remains the same throughout the entire series, as can be seen on the figures. One possible explanation to the pronounced displacement of Ha is the preferred conformation these molecules may adopt. Once the chiral auxiliaries are introduced to the corresponding carboxylic acids, the molecules adopt an anti-s-cis conformation. In this conformation, Ha experiences the proximity of the oxygen carbonyl or the sulfur thionyl group of the chiral auxiliary, where the deshielding effect of these groups induces the downfield displacement of Ha. This type of displacement experienced by Ha when the auxiliaries are introduced is observed throughout all the series, thus supporting the idea of an anti-s-cis conformation as the most stable conformation for all these compounds. (Fig. 4 and 5) In addition to the 1H NMR evidence, the anti-s-cis conformational preferences shown by these systems may be the result of some C-Ha...O or C-Ha...S hydrogen bond in a cyclic six-membered ring arrangement [11].
Computational studies
We have performed electronic structure calculations within the Density Functional Theory (DFT) in order to determine the energies of all the possible conformers appearing in 8-10. In particular we used the hybrid B3LYP exchange-correlation functional [12] with the standard 6-31G** basis sets for all atoms. Geometry optimizations for the four conformers of each species were carried out without any symmetry restrictions and the nature of the minima was verified with analytical frequency calculations (no imaginary frequencies) for all the conformers studied here. Gibbs free energies were obtained at T = 298.15 K within the harmonic approximation. These optimizations considered up to 162 degrees of freedom. In the present case all of the critical degrees of freedom are dihedral angles. The calculations were done using the Gaussian03 code [13]. The default code-supplied thresholds (maximum and RMS force/displacements) were used for the convergence criteria.
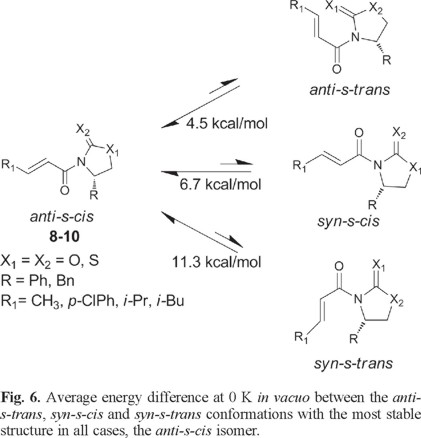
Based on the results found with the NMR experiments, we now turn our attention to the relative stability of the four possible conformers in each case. Table 2 shows the relative the B3LYP/6-31G* energies of all compounds for the four different conformers of each species obtained at 0 K in vacuo. We find that, in all cases, the anti-s-cis conformation is the most stable one compared to the other three isomers for each series of molecules (compounds 8-10). Another important result is that, at 0 K and without solvent effects, the relative stability of these conformers is anti-s-cis > anti-s-trans > syn-s-cis > syn-s-trans and the mean relative energies between the four type of conformers are 4.5 (anti-s-cis/anti-s-trans), 2.2 (anti-s-trans/syn-s-cis) and 4.6 (syn-s-cis/syn-s-trans) kcal/mol.
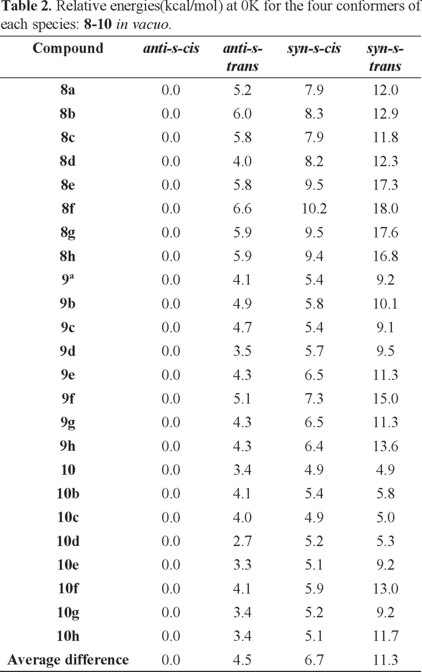
Regarding the benzyl substituent in compounds 8e and 8f, a conformation in which this substituent is oriented further apart from the double bond and the chiral auxiliaris ring systems was considered for the calculations. In all cases, this orientation resulted in the lower energy conformation.
At this point it is interesting to note that the average energy difference in vacuo between the most stable conformation (anti-s-cis) and the conformation generally used to illustrate the structure and reactivity of N-enoyl systems (syn-s-cis) [2] is 6.7 kcal/mol and, quite remarkably, the latter is not even the second most stable conformer (see Fig. 6).
In order to study the role of finite temperature on the relative stability of the conformers, we have performed a set of Gibbs free energy calculations at 298.15 K for the four conformers of compounds 8a, 8e, 9a, 9e, 10a and 10e. Table 3 shows a comparison of the relative energies using the internal energy ΔE at 0 K and those obtained including the ZPE and entropic contributions at 298 K, ΔG298.
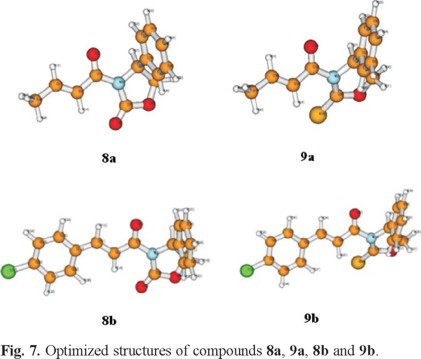
Note that the largest change with respect to the energy differences at 0 K between the two most stable conformers is < 0.5 kcal/mol. The important result derived from Table 3 is that the stability order established previously in Table 2 for the four conformers is not modified when considering the Gibbs free energy differences in vacuo at room temperature. Fig 7 shows the optimized structures of compounds 8a, 9a, 8b and 9b compounds whose 1H NMR spectra are given above. The fact that the anti-s-cis conformers are the most stable structures in all cases is in good agreement with the experimentally observed 1H NMR chemical shifts. The optimized geometries for all isomers of each species are available upon request from the authors.
Finally, since the NMR spectra were obtained in chloroform, we deemed necessary to verify if the free energy differences are modified by the solvent, and if the solvent effects are large enough to qualitatively change the stability order of the conformers. In addition, we also considered toluene since this solvent is used in our medicinal chemistry program. Therefore, for the 8a and 8e cases, the Gibbs free energy differences at 298.15 K between the four conformers were also calculated using the Polarizable Continuum Model (PCM) [14] using chloroform and toluene as solvents. The inclusion of the solvent effects in the PCM scheme is done via their dielectric constants. As previously done in vacuo, full geometry optimizations and frequency calculations were done considering the solvent effects. We note that the final optimized geometries in the solvents were barely modified from those optimized in vacuo. The results of these calculations are presented in Table 4.
Table 4 shows that the inclusion of the solvent effects, for both solvents, diminishes the free energy differences between the conformers; nevertheless, their relative stability is the same as that obtained in vacuo. As a byproduct we also find that chloroform has a larger effect on the energy differences among the four conformers of each species than toluene.
In particular, this comparison shows that the energies of the lowest lying conformers, the anti-s-cis and anti-s-trans structures, are lowered in the solvent by roughly the same amount (< 1 kcal/mol), so that the energy differences in vacuo yield a good approximation to the Gibbs free energy differences in solution at 298 K. For instance, in the case of 8a the free energy difference between the anti-s-cis and the anti-s-trans conformers at 298 K in chloroform is 4.4 kcal/mol, as compared to the 4.8 kcal/mol value obtained in vacuo; for the 8e case the free energy difference between the anti-s-cis and the anti-s-trans conformers at 298 K in chloroform is 5.4 kcal/mol, as compared to the 5.6 kcal/mol value obtained in vacuo. Therefore, these electronic structure calculations confirm the greater stability of the anti-s-cis conformers in all cases, both in vacuo and when considering the finite temperature and solvent effects.
Conclusion
In conclusion, an anti-s-cis 2 conformation is consistent with the 1H NMR spectra of these compounds where the proximity of the carbonyl or thionyl group of the chiral auxiliaries induces a high frequency chemical shift of the vinylic proton Ha. On the other hand, the systematic electronic structure calculations carried out for the N-enoyl systems 8a-h to 10a-h confirm the anti-s-cis conformation as the most stable one. The stability order found in vacuo for the conformers (anti-s-cis > anti-s-trans > syn-s-cis > syn-s-trans) is conserved if solvent effects in chloroform and toluene are included. The anti-s-cis 5 conformer is, on average, ca. 6 kcal/mol more stable than the one usually assumed (syn-s-cis 4) to explain the reactivity and stereochemistry of these compounds.
Acknowledgments
MFZ and ARS gratefully acknowledge financial support from CONACYT (México) through basic science projects No. 82129 and 130931. ARS also thanks sabbatical support from the French NEXT and INSA-Toulouse visiting scholar programs.
References
1. (a) Alexakis, A.; Bäckwall, J.E.; Krause, N.; Pamies, O.; Dieguez, M. Chem. Rev. 2008, 108, 2796-2823. [ Links ] (b) Harutyunyan, S.R.; Hartog, T.; Geurts, K.; Minnard, A. J.; Feringa, B.L. Chem. Rev. 2008, 108, 2824-2852. [ Links ] (c) López, F.; Minnaard, A.J.; Feringa, B.L. Acc. Chem. Res. 2007, 40, 179-188. [ Links ] (d) Fleming, F.F.; Wang, Q. Chem. Rev. 2003, 103, 2035-2077. [ Links ] (e) Krause, N; Hoffmann-Röder, A. Synthesis 2001, 171-196. [ Links ]
2. (a) Williams, D.R.; Nold, A.L.; Mullins, R.J. J. Org. Chem. 2004, 69, 5374-5382. [ Links ] (b) Esumi, T.; Shimizu, H.; Kashiyama, A.; Sasaki, C.; Toyota, M. Tetrahedron Lett. 2008, 49, 6846-6849. [ Links ] (c) Morita, M.; Ishiyama, S.; Koshino, H.; Nakata; T. Org. Lett. 2008, 10, 1675-1678. [ Links ] (d) Williams, D.R.; Ihle, D.C.; Brugel, T.A. Patnaik, S. Heterocycles 2006, 70, 77-82. [ Links ]
3. (a) Castellino, S.; Dwight, W.J. J. Am. Chem. Soc. 1993, 115, 2986-2987. [ Links ] (b) Laszlo, P., Teston. M. J. Am. Chem. Soc. 1990, 112, 8750-8754. [ Links ] (c) Evans, D.A.; Chapman, K.T; Bisaha, K. T. J. J. Am. Chem. Soc. 1988, 110, 1238-1256. [ Links ]
4. Williams, D.R.; Kissel, W.S.; Li, J.J. Tetrahedron Lett. 1998, 39, 8593-8596. [ Links ]
5. (a) Dambacher, J.; Anness, R.; Pollock, P.; Bergdahl, M. Tetrahedron 2004, 60, 2097-2110. [ Links ] (b) Polock, P.; Dambacher, J.; Anness, R.; Bergdahl, M. Tetrahedron Lett. 2002, 43, 3693-3697. [ Links ]
6. Sabala, R.; Hernández-García, L.; Ortiz, A.; Romero, M.; Olivo. H.F. Org. Lett. 2010, 12, 4268-4270. [ Links ]
7. Lu, C.-F.; Zhang, S.-B.; Li, Y.; Yang, G.-G.; Chen, Z.-X. Tetrahedron: Asymmetry 2009, 20, 2267-2269. [ Links ]
8. Cardillo, G.; Gentilucci, L.; Gianotti, M.; Tolomell, A. Org. Lett. 2001, 3, 1165-1167. [ Links ]
9. (a) Andrade, C.K.Z.; Rocha, R.O.; Vercillo, O.E.; Silva, W.A.; Matos, R.A.F. Synlett 2003, 2351-2352. [ Links ] (b) Nicolás, E.; Russell, K.C.; Hruby, V.J. J. Org. Chem. 1993, 58, 766-770. [ Links ] (c) Williams, D.R.; Turske, R.A. Org. Lett. 2000, 2, 3217-3220. [ Links ] (d) Palomo, C.; Oiarbide, M.; Lopez, R.; Gonzalez, P.B.; Gomez-Bengoa, E.; Saa, J.M.; Linden, A. J. Am. Chem. Soc. 2006, 128, 15236-15247. [ Links ] (e) Munive, L.; Rivas, V.M.; Ortiz, A.; Olivo, H.F. Org. Lett. 2012, 14, 3514-3517. [ Links ] (f) Bellassoued, F.; Lensen, N.; Bakasse, M.; Mouelhi, S. J. Org. Chem. 1998, 63, 8785-8789. [ Links ]
10. (a) Davies, S.G.; Fletcher, A.M.; Hermann, G.J.; Poce, G.; Roberts, P.M.; Smith, A.D.; Sweet, M.J.; Thomson, J.E. Tetrahedron:Asymm 2010, 21, 1635-1648. [ Links ] (b) Evans, D.A.; Chapman, K.T.; Bisaha, J. J. Am. Chem. Soc. 1988, 110, 1238-1256. [ Links ]
11. (a) Sigalov, M.; Krief, P.; Shapiro, L.; Khodorkovsky, V. Eur. J. Org. Chem. 2008, 673-683. [ Links ] (b) Wong, M.W. J. Org. Chem. 2005, 70, 5487-5493. [ Links ]
12. (a) Becke, A.D. Phys. Rev. A. 1988, 38, 3098-3100. [ Links ] (b) Lee, C.; Yang, W.; Parr, R.G. Phys. Rev. B. 1988, 37, 785-789. [ Links ]
13. Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A. Jr.; Stratmann, R.E.; Burant, J.C.; Dapprich, S.; Millam, J.M.; Daniels, A.D.; Kudin, K.N.; Strain, M.C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G.A.; Ayala, P.Y.; Cui, Q.; Morokuma, K.; Salvador, P.; Dannenberg, J.J.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, B.; Cioslowski, J.; Ortiz, J.V.; Baboul, A.G.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.; Gill, P.M.W.; Johnson, B.; Chen, W.; Wong, M.W.; Andres, J.L.; Gonzalez, C.; Head-Gordon, M.; Replogle, E.S.; Pople, J.A. Gaussian 03 (Gaussian, Inc., Pittsburgh, PA, 2003). [ Links ]
14. (a) Tomasi, J.; Mennucci, B.; Cammi, R. Chem. Rev. 2005, 105, 2999-3093. [ Links ] (b) Tomasi, J.; Mennucci, B.; Cancès. E. J. Mol. Struct. (THEOCHEM). 1999, 464, 211-226. [ Links ]

