










Services on Demand
Journal

Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
 Cited by SciELO
Cited by SciELO Related links
 Similars in
SciELO
Similars in
SciELO Share

 Permalink
PermalinkJournal of the Mexican Chemical Society
Print version ISSN 1870-249X
J. Mex. Chem. Soc vol.58 n.1 Ciudad de México Jan./Mar. 2014
Article
Liquid Chromatography for the Analysis of Hydrophilic Drugs in the Presence of Ionic Liquids
Marsela Garza Tapia,1 Abelardo Chávez Montes,2 Encarnación Moyano Morcillo,3 Ma. Teresa Galceran i Huguet,3 Noemí H. Waksman de Torres,1 and Rocío Castro Ríos1*
1 Departamento de Química Analítica, Facultad de Medicina, Universidad Autónoma de Nuevo León, Av. Madero s/n, Mitras Centro, 64460, Monterrey, Nuevo León, México, rcastro_r@yahoo.com.mx
2 Departamento de Química, Facultad de Ciencias Biológicas, Universidad Autónoma de Nuevo León, Av. Pedro de Alba s/n, Cd. Universitaria, 66451, San Nicolás de los Garza, Nuevo León, México.
3 Departamento de Química Analítica, Facultad de Química, Universidad de Barcelona, Martí i Franquès, 1-11, 08028, Barcelona, España.
Received January 31, 2013.
Accepted September 17, 2013.
Abstract
Currently available methods for the analysis of hydrophilic compounds are usually time-consuming, tedious and inefficient. Ionic liquids (ILs) are salts with low melting points that are described as designer solvents. When ILs are used in the extraction of hydrophilic compounds, liquid chromatographic analysis can be challenging, as a result of co-elution of the IL with the analyte and poor miscibility of the IL with the mobile phase. This paper describes the development of analytical methods for hydrophilic compounds in the presence of water-immiscible ILs. Conventional and ion-pair reversed phase and hydrophilic interaction liquid chromatography were tested, with the best results obtained with the last one. The proposed methods showed a good performance and represent a useful alternative for the analysis of hydrophilic drugs.
Keywords: Ionic liquids, HPLC, hydrophilic interaction chromatography, drugs, hydrophilic compounds.
Resumen
Los métodos actualmente disponibles para el análisis de compuestos hidrofílicos son, en general, largos, tediosos y poco eficientes. Los líquidos iónicos (LIs), son sales con puntos de fusión bajos, considerados como disolventes de diseño. Cuando los LIs se emplean en la extracción de compuestos hidrofílicos, el análisis por cromatografía líquida puede convertirse en un reto, por problemas de coelución y de miscibilidad de con la fase móvil. Este trabajo presenta el desarrollo de métodos de análisis para compuestos hidrofílicos en presencia de LIs inmiscibles en agua, se evaluaron la cromatografía en fase inversa convencional y de par iónico, así como, la cromatografía de interacciones hidrofílicas, siendo esta última la que mostró mejores resultados. Los métodos propuestos presentaron un buen desempeño y representan una alternativa útil para el análisis de fármacos hidrofílicos.
Palabras clave: Líquidos iónicos, CLAR, cromatografía de interacciones hidrofílicas, fármacos, compuestos hidrofílicos.
Introduction
In recent years, the analysis of hydrophilic compounds has attracted an increasing interest as such compounds are widely employed in the manufacture of a number of products commonly used at home and industry, including drugs, pesticides, and personal care products [1]. Due to their wide distribution, many of these substances are also considered to be emerging environmental contaminants.
However, the analysis of hydrophilic compounds is often difficult. Lengthy and tedious extraction processes consume large volumes of solvents, yet these methods have poor yields because of the low affinity of hydrophilic compounds for conventional organic solvents.
Ionic liquids (ILs) are non-molecular solvents that have recently gained considerable attention as a new class of designer solvents [2-4]. These compounds are salts with melting points below room temperature [3] and are composed of bulky and asymmetric organic cations with various anions. The anions are usually inorganic but can also be organic [5], and they are primarily responsible for the miscibility of ILs with water [4]. Among the main features of ILs are a low vapor pressure, high thermal [2-4] and oxidative [4] stability, and unique catalytic properties [2]. Because of their low volatility, the ILs are considered “green solvents” with respect both to the safety of the operator and to the environment [4, 6]. This characteristic makes the ILs a potentially attractive replacement for volatile organic compounds as the solvent in various chemical processes. Another important advantage of ILs is the ability to adjust their physicochemical properties by the choice of the cation and anion, providing a wide range of potential solvents [5, 3]; this last feature makes these compounds extremely interesting for extraction processes.
The usefulness of ILs has been demonstrated in liquid-liquid microextraction, microwave-assisted extraction, dispersive liquid-liquid microextraction [7], single-drop microextraction [8,9], and solvent bar microextraction [6] among others. Nevertheless, subsequent analysis by liquid chromatography can be challenging, due to the high viscosity of ILs, its immiscibility with common mobile phases and co-elution particularly with hydrophilic analytes. In spite of these problems, only few reports on the IL compatibility of analytical methods have been published [10].
ILs have been used in liquid chromatography as both mobile phase modifiers [11-13] and structural components of the stationary phase [10, 13-15]. Also, some work has been done on the study of the interaction of ILs with various stationary phases and their potential retention mechanisms, mainly using reversed-phase high performance liquid chromatography (RP-HPLC) [16, 17]. It has been shown that in RP-HPLC, mobile phases with large aqueous fractions decrease miscibility of certain ILs, especially those with bis(triluoromethylsulfonyl)imide anions [18]. In the other hand, the use of columns with mixed stationary phases or hydrophilic interaction liquid chromatography (HILIC) can provide better retention and selectivity for ILs. Besides, the use of mobile phases containing a large fraction of organic solvent is also helpful because it improves the miscibility of certain ionic liquids and facilitates the use of chromatography in conjunction with such techniques as mass spectrometry [18]. However, most reports dealing with the separation of ionic liquids from other compounds after their use as extraction solvents have focused on non polar or moderately polar analytes, such as aromatic compounds and pesticides [7, 9, 20, 21]; there are few reports for polar or hydrophilic analytes [22, 23]. It is worth noting that the majority of these reports do not address whether ILs affect chromatographic separations or pose other difficulties for proper identification and quantification of analytes; in fact, many these works do not even present any chromatogram. Our research group has observed an important influence on chromatography when ILs are present. This behavior has also been reported by Berton et al. [24] while using ionic liquids as extraction solvents. These researchers observed a significant variation in analyte retention when the sample contained an ionic liquid [24].
Our work with ILs is mainly focused to the development of green and efficient liquid-phase microextraction procedures for the analysis of hydrophilic compounds. In this paper, the development of chromatographic methods for the analysis of hydrophilic drugs in the presence of water-immiscible ILs is presented and the difficulties found in the chromatographic analysis of IL-containing matrices are discussed. As model analytes some hydrophilic drugs were included in this work, while ILs were selected from the commonly used in the microextraction methods above mentioned. The studied water-inmiscible ILs were composed of organic cations such as imidazolium, pyridinium, pyrrolidinium, pyperidinium, sulfonium and ammonium; whereas the anions included were hexafluorophosphate, bis(trifluoromethylsulfonyl)imide, tetrafluoroborate and trifluoromethanesullfonate.
Results and Discussion
RP, Ion-pair, and HILIC chromatographies were tested for the separation of ILs from hydrophilic drugs, including. Likewise, the performance of different columns was evaluated within these types of chromatography. The separation conditions were established first by injecting each analyte dissolved in the appropriate solvent. After that, the behavior of ILs in all the developed chromatographic methods was subsequently evaluated by analyzing individual solutions of each IL.
For reversed-phase chromatography, the separation was evaluated initially using two columns (Atlantis dC18 and Zor-bax Eclipse XDB C18) and the mobile phases described in Experimental section. The Atlantis column has been manufactured for use with highly aqueous mobile phases and is recommended for polar compound retention, whereas the Zorbax one has a deactivated stationary phase that can work over a wide pH range (2 to 9) and, in accordance to the manufacturer's specifications, provide more symmetrical chromatographic peaks, particularly for basic compounds.
With both columns and all mobile phases, retention of analytes was achieved. Although, the Zorbax column gave shorter retention times in all cases and some compounds eluted near to the column dead time. For most analytes no significant influence of pH was found and no differences neither for retention times nor for peak shapes were observed. Only in the case of salycilic acid, when using mobile phases with pH values of 3 too high retention times and wide peaks were obtained. So a pH 4 formate-formic acid buffer solution (100 mM) was selected for further work. In order to get a satisfactory retention, the organic modifier content was changed between 5 and 30%, working always in isocratic mode. Table 1 summarizes the best chromatographic conditions for each individual drug on the Atlantis column. It must be noted that neither of these columns allowed simultaneous separation of the analytes.

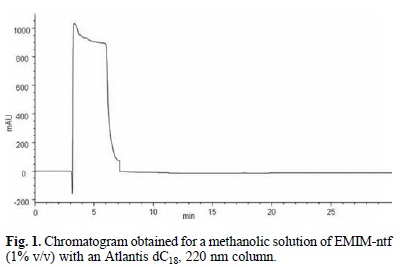
Next, having established the chromatographic conditions for each analyte, the behavior of the ILs was studied by analyzing methanolic solutions (1% v/v) of each IL. It most be noted that the 1% concentration level used in this experiments was chosen keeping in mind that almost always samples from microextraction must be diluted with an adequate solvent before injection due to the immiscibility and/or viscosity of the extraction solvents. In our case, most of the ILs showed wide signals that eluted at same time as the model drugs included in the study. For instance, Fig. 1. shows the chromatogram obtained for EMIM-ntf under the conditions established for caffeine. As it can be observed, a poor signal shape was attained and this was attributed to the high content of methanol in the sample. Nevertheless, the use of solvents with higher aqueous content, compatible with the mobile phase use was not possible due to the immiscibility of the ILs.
So, a Discovery HS F5 column with pentafluorophenyl-functionalized stationary phase was evaluated. According to the manufacturer, this column provides increased retention times of analytes with log Ko/w values lower than 2.5 and a more efficient separation of compounds with similar retention times in C18 columns. Various mobile phase compositions were tested with this column, and using a gradient it was possible to obtain a method for the simultaneous separation of the hydrophilic drugs, as shown in Fig. 2. Nevertheless, although under these conditions there were no peaks from the ILs that interfered with the analytes, the required use of methanol as a solvent to facilitate the miscibility of ILs caused a loss of resolution and signal distortion of analytes, as shown in Fig. 3. As with RP-HPLC, It became impossible to get a homogeneous solution of the ILs and the mobile phase, even though numerous attempts were made.
Taking into account the ionic properties of ILs, ion-pair chromatography was considered next with the goal of better retaining the ILs and thus differentiating their signals from those of the drugs. However, this type of chromatography also proved to be inefficient for separation because, as observed in RP-HPLC, many ionic liquids showed signals in the drug-eluting range.
Considering the problems described above, HILIC was evaluated as an alternative due to its ability to separate hydrophilic compounds and its use of a large organic fraction in the mobile phase, which appeared suitable for the water-immiscible ionic liquids. Two columns were tested with this type of chromatography: an Atlantis HILIC Silica and a Luna CN-, which exhibited no significant differences in their retention of the analytes, however it was possible to retain only atenolol, cimetidine, metformin and ranitidine,as the rest of analytes exhibited notably low retention times (< 1.5 min), virtually eluting with dead time. HILIC is commonly recommended for analysis of highly polar compounds, nevertheless the separation mechanisms are currently not well understood. Thus, it may be difficult to predict the behavior of compounds in this chromatographic mode. In this case, we observed that increasing the buffer concentration from 250 to 500 mM produced a slightly enhancement on the retention, so to obtain a satisfactory retention time and separation with all ILs, a very high ionic strength would be necessary.
A pH 5 aqueous mobile phase was selected as analytes showed higher retention times. Regarding the effect of the ionic strength of aqueous fraction it was observed that higher salt concentration yielded narrower peaks. In the same way as RP-HPLC, the acetonitrile content in mobile phase was tested in order to obtain good retention and shape of the chromatographic signals. Table 2 shows the best conditions and retention times for each of the drugs using HILIC.
ILs were tested using the HILIC proposed methods. It was possible to get an adequate separation of drugs and most of the ILs, because the latter usually showed no retention, and those that were retained did not interfere with the analytes, in Table 2 ILs for which no separation from the drugs was achieved are also listed.
The linearity, precision, accuracy, and the limits of detection and quantification were evaluated to validate each of the established HILIC methods. Table 3 provides a summary of the results of these validation parameters for each method. Standard solutions were prepared for each drug in triplicate at five concentration levels between 5 and 80 ppm to generate calibration curves. A linear fit was performed by least-squares regression, and all methods were found to be linear with r2 values> 0.99. The precision was evaluated by the relative standard deviation of the response factors of the calibration standards, which, in all cases, was less than 5%. To assess the accuracy of the established methods, the correlation between actual and expected concentrations of the calibration standards was evaluated and, for all analytes, values of r2 > 0.99 and m around 1 were obtained. Finally, the limits of detection (LOD) and quantification (LOQ) were calculated using a signal-to-noise ratio of 3 and 10, respectively. The LOD values thus obtained ranged from 0.25 to 0.84 ppm, and the LOQ values ranged from 0.78 to 2.55 ppm. These results showed an adequate performance of the methods. At our knowledge, no similar studies dealing with the analysis of hydrophilic drugs in ILs-containing matrices have been published until now but the proposed methods can be considered a good alternative to the available ones as it overcomes the drawbacks of the use of normal phase [25], ion-pair reagents [26], analyte derivatization [27].
Materials and Methods
Reagents
Pure standards were used for R-atenolol (99%), caffeine (Reagent Plus), cimetidine, furosemide, famotidine, hydrochlorothiazide, L-dopa, metformin (1,1-dimethylbiguanide hydrochloride, 97%), acetaminophen (99%), ranitidine hydrochloride, salicylic acid (>99%) all of which were purchased from Sigma-Aldrich (USA). Acetonitrile and methanol (JT Baker, USA), both HPLC grade, were used for the preparation of the mobile phase along with trifluoroacetic (99%) and heptafluorobutyric (99%) acids and ammonium hydroxide (25%) from Sigma-Al-drich (USA), formic acid (99%, Merck, Germany), and acetic acid (99.8%, Fluka, USA). Deionized water was obtained with an Elga II system (Veolia, Mexico).
The ionic liquids were purchased from Iolitec (Germany) and included: 1-butyl-3-methylimidazolium hexafluorophos-phate (BMIM-PF6), 1-ethyl-3-methylimidazolium bis(triflu oromethylsulfonyl)imide (EMIM-NTf), 1-hexyl-3-methyl-imidazolium bis(trifluoromethylsulfonyl)imide (HMIM-NTf), 1-hexyl-3-methylimidazolium hexafluorophosphate (HMIM-PF6), 1-hexyl-3-methylimidazolium trifluoromethanesulfonate (HMIM-OTf), 1-octyl-3-methylimidazolium hexafluorophosphate (OMIM-PF6), 1,2-dimethyl-3-propylimidazolium bis(tr ifluoromethylsulfonyl)imide (DMPIM-NTf), 3-methyl-1-pro-pyl-pyridinium bis(trifluoromethylsulfonyl)imide (PMPI-NTf), trimethyl(butyl)ammonium bis(trifluoromethylsulfonyl)imide (N1114-NTf), methyl(trioctyl)ammonium bis(trifluoromethyls ulfonyl)imide (N1888-NTf), 1-methyl-1-propyl-pyrrolidinium bis(trifluoromethylsulfonyl)imide (BLP-NTf), 1-methyl-1-propyl-piperidiniumbis(trifluoromethylsulfonyl)imide (MP-PIP-NTf), triethylsulfonium bis(trifluoromethylsulfonyl)imide. (SEt3-NTf), and 1-decyl-3-methylimidazolium tetrafluoroborate (bmim-BF4).
Chromatographic analysis
For the development of the chromatographic methods, a 2695 Liquid Chromatography system (Waters, USA) equipped with an online degasser, a quaternary pump, an autosampler, a column oven, and a diode array detector was employed. We also used an HP 1100 series (Hewlett-Packard, USA) instrument with an online degasser, a quaternary pump, an autosampler, a column oven, and a UV variable wavelength detector. The wavelengths used for the analysis of each of the analytes were as follows: acetylsalicylic acid, acetaminophen, and caffeine, 254 nm; atenolol, famotidine, furosemide, hydrochlorothiazide, L-dopa, and ranitidine, 280 nm; cimetidine and metformin, 220 and 230 nm, respectively.
The columns evaluated in the study were: Atlantis dC18 (2.1 x 150 mm, 3 pm; Waters, USA), Zorbax Eclipse XDB C18 (2.1 x 150 mm, 5 pm; Agilent, USA ), Discovery HS F5 (2.1 x 150 mm, 3 pm, Supelco, USA), Kromasil C8 (2.1 x 150 mm, 5 pm; Phenomenex, USA), Atlantis HILIC Silica (2.1 x 150 mm, 5 pm; Waters, USA) and Luna CN- (2.0 x 150 mm, 3 pm; Phenomenex, USA).
For RP-HPLC, mixtures of methanol with 100 mM for-mate/formic acid buffer solution (pH 3 and 4) were tested as mobile phases, to evaluate the effects of pH and organic modifier content (5, 10, 20 and 30%). For Ion-pair HPLC, we used trifluoroacetic acid- and heptafluorobutyric acid-water solutions (20 mM, pH 4) mixed with methanol or acetonitrile (10-30%). For HILIC, we used mobile phases with high acetonitrile content (80-95%) mixed with 40, 100 and 250 mM acetate/acetic acid buffer solutions (pH 4 and 5). In each case, the mobile phase flow rate was established by considering its composition and the characteristics of the column. Separations were carried out at 30 °C or 40 °C and mobile phase flow-rates were established taking into account column dimensions and back pressures.
Method development was carried out using aqueous, methanolic, or acetonitrile solutions for the analytes (20 ppm) and ionic liquids (1% v/v) according to the mobile phase composition; in all cases 10 pL were injected. All experiments were performed in duplicate.
Conclusions
In this work, HPLC methods have been developed for the determination hydrophilic drugs in ILs-containing matrices. RP-HPLC, in the conventional and in ion-pair modes, presented some limitations to achieve a satisfactory separation as coelution occurred. Moreover, the immiscibility of ionic liquids with aqueous mobile phases made it necessary to dilute the samples using the organic solvents, producing peak distortions. In the other hand, the use of HILIC allowed to resolve some of the analytes from the ILs. Proposed methods proved to be accurate and precise and a good alternative for the analysis of hydrophilic drugs.
References
1. Quitana, J. B.; Rodriguez, I.. Anal. Bioanal. Chem. 2006, 384, 1447-1461. [ Links ]
2. Lopez-Darias, J.; Pino, V.; Anderson, J. L.; Graham, C. M.; Afonso, A. M.. J. Chromatogr. A. 2010, 1217, 1236-1243. [ Links ]
3. Liu, R.; Liu, J. F.; Yin, Y. G.; Hu, X. L.; Jiang, G. B. Anal. Bioanal. Chem. 2009, 393, 871-883. [ Links ]
4. Baker, G. A.; Baker, S. N.; Pandey, S.; Bright, F. V. Analyst 2005, 130, 800-808. [ Links ]
5. Marcilla, R.; Mecerreyes, D. An. Real Soc. Esp. Quím. 2005, 22-28. [ Links ]
6. Guo, L.; Lee, H. K. J. Chromatogr. A 2011, 1218, 4299-4306. [ Links ]
7. Wang, X.; Xu, Q. C.; Cheng, Ch. G.; Zhao, R. S. Chromatographia 2012, 75, 1081-1085. [ Links ]
8. Sheikhloie, H.; Saber-Tehrani, M.; Abrumand-Azur, P.; Waqif-Husain, S. Acta Chromatogr. 2009, 4, 577-589. [ Links ]
9. Yao, C.; Pitner, W. R.; Anderson, J. L. Anal. Chem. 2009, 81, 5054-5063. [ Links ]
10. Wahlstrom, R.; Rovio, S.; Suurnakki, A. Carbohydrate Res. 2013, 373, 42-45. [ Links ]
11. Cruz-Vera, M.; Lucena, R.; Cárdenas, S.; Valcárcel, M. Anal. Bioanal. Chem. 2008, 391, 1139-1145. [ Links ]
12. Herrera-Herrera, A. V.; Hernández-Borges, J.; Rodríguez-Delgado, M. A. Anal. Bioanal. Chem. 2008, 392, 1439-1446. [ Links ]
13. Sun, P.; Armstrong, D. W. Anal. Chim. Acta 2010, 661, 1-16. [ Links ]
14. Van Meter, D. S.; Oliver, N. J.; Carle, A. B.; Dehm, S.; Ridgway, T. H.; Stalcup, A. M. Anal. Bioanal. Chem. 2009, 393, 283-294. [ Links ]
15. Wang, Y.; Zhu, T.; Ho-Row, K. J. Chromatogr. Sci. 2010, 48, 690-693. [ Links ]
16. Nichthauser, J.; Stepnowski, P. J. Chromatogr. Sci. 2009, 47, 247-253. [ Links ]
17. Stepnowski, P.; Nichthauser, J.; Mrozik, W.; Burszewski, B. Anal. Bioanal. Chem. 2006, 385, 1483-1491. [ Links ]
18. Lamoroux, C.; Foglia, G.; Le Rouzo, G. J. Chromatogr. A 2011, 1218, 3022-3028. [ Links ]
19. Chang Fan, Y.; Liang Hu, Zh.; Lan Chen, M.; Shen Tu, Ch.; Zhu, Y. Chin. Chem. Lett. 2008, 19, 985-987. [ Links ]
20. Bai, H.; Zhou, Q.; Xie, G.; Xiao, J. Anal. Chim. Acta 2009, 651, 64-68. [ Links ]
21. He, L.; Luo, X.; Xie, H.; Wang, CH., Jiang, X.; Lu, K. Anal. Chim. Acta 2009, 655, 52-69. [ Links ]
22. Mizuuchi, H.; Jaitely, V.; Murdan, S.; Florence, A. T. Eur. J. Pharm. Sci. 2008, 33, 326-331. [ Links ]
23. Yao, C.; Li, T.; Twu, P.; Pitner, W. R.; Anderson, J. L. J. Chromatogr. A 2011 1218, 1556-1566. [ Links ]
24. Berton, P.; Monasterio, R. P.; Wuilloud, R. G. Talanta 2012, 97, 521-526. [ Links ]
25. Mante-Sarcero, S.; Sierra, I. Chirality 2012, 24, 860-866. [ Links ]
26. Helali, N.; Monser, L. Chromatographia 2006, 63, 425-430. [ Links ]
27. Önal, A. Eur. J. Med. Chem. 2009, 44, 4998-5005. [ Links ]

