










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkJournal of the Mexican Chemical Society
Print version ISSN 1870-249X
J. Mex. Chem. Soc vol.56 n.1 Ciudad de México Jan./Mar. 2012
Article
Chromatin Bound PCNA is Complexed with Cell Cycle Protein Regulators as Determined by Chromatin Immunoprecipitation
Nazul Becerril,1 Miguel Ángel Martínez,1 Elpidio García, and Jorge Manuel Vázquez Ramos*
Facultad de Química, Departamento de Bioquímica, Universidad Nacional Autónoma de México. Avenida Universidad y Copilco, México D.F. 04360, México. 01 55 56225284, jorman@unam.mx
Received January 27, 2011.
Accepted April 6, 2011.
Abstract
Using the technique of Chromatin Immunoprecipitation, we have detected the formation of complexes between the homotrimer protein ring Proliferating Cell Nuclear Antigen, PCNA, and two fundamental regulators of the cell cycle, CdkA and Cyclin D4;2 along germination of maize seeds. This finding suggests the way PCNA participates in the control of the G1 phase, by allowing cyclin–kinase complexes to find and phosphorylate protein targets. Incidentally, PCNA may form a structure of a dimer of trimer when bound to the chromatin.
Key words: Maize, PCNA, D cyclins, Cdks, ChIP, cell cycle.
Resumen
Mediante inmunoprecipitación de la cromatina, hemos detectado la formación de complejos entre la proteína homotrimérica anular Antígeno Nuclear de Proliferación Celular, ANPC, y dos reguladores fundamentales del ciclo celular, CdkA y Ciclina D4;2 durante la germinación de maíz. Se sugiere la forma en que ANPC participaría en el control de la fase G1, al permitir al complejo ciclina–cinasa encontrar y fosforilar proteínas blanco. Incidentalmente, ANPC podría formar una estructura de dímero de trímero cuando se halla unida a la cromatina.
Palabras clave: Maíz, ANPC, ciclinas D, Cdks, ChIP, ciclo celular.
Introduction
The Proliferating Cell Nuclear Antigen (PCNA) was initially found as a nuclear protein that was recognized by auto–antibodies in patients with systemic lupus erythematosus and whose synthesis was related with a proliferative state of the cells [1] since it was synthesized during the S phase of the cell cycle. PCNA is a highly conserved protein, with minimal differences in yeasts, mammals, humans or plants [2–4] and a conserved molecular mass of around 29 kDa. Crystallography studies have shown that PCNA is a homotrimer forming a hexagonal ring. Each monomer contains a region called InterDomain Connector Loop (IDCL) which is bound by different proteins possessing a domain known as of Interaction to PCNA or PIP.
PCNA is essential for DNA metabolism, playing fundamental roles in replication, repair, recombination and also in cell cycle control [5–7]. It was initially described as a protein factor that stimulated activity and increased processivity of δ and ε–type DNA polymerases [8–10], acting as a "sliding clamp" on DNA in a Replication Factor C (RF–C)–dependent way to allow the entry of the replicative DNA polymerase to the replication fork, stimulating it.
Accumulating evidence indicates that, in mammal cells, PCNA associates to an increasing number of proteins that participate in DNA metabolism, as DNA polymerases S and e, RFC, Fen1, DNA ligase 1, DNA topoisomerase, MLHI, MSH2, XP–G, DNA methyl–transferases and many others [1016]. Thus, it has been proposed that PCNA, besides being a processivity factor for DNA polymerases, it works as a recruiting platform for these and other proteins to the replication fork, or else to DNA with single stranded fragments, indicative of DNA damage [17].
PCNA also associates to proteins functioning in cell cycle control, as D–cyclins, cyclin–dependent kinases (Cdks) and p21 [7, 18, 19]. Cyclin D is a protein that binds and activates a Cdk [20]. Cdks are a family of protein kinases whose function is to regulate the cell cycle and p21 is a regulator of cyclin–Cdk kinases. PCNA has been found forming a quaternary complex with cyclin D, Cdk and p21. The physiological meaning of this complex has not been determined, but it is suggested that it may contribute to coordination between cell cycle progression and DNA replication [21, 22].
Research in our group has been focused in the study of the G1 phase and the G1–S transition of the cell cycle and its relationship with a developmental process such as maize seed germination and, for this purpose, it has been focused to the study of some of the proteins that determine entry into the G1 phase such as the D–type cyclins and the A–type Cdks [23, 24], and also in a fundamental protein for S phase, PCNA. Independently of its S–phase function, and similarly to what it has been found in mammal cells, during maize germination, PCNA also associates to D–type cyclins and to A–type Cdks [25]. These ternary complexes show CdkA–type kinase activity and this activity seems to be more relevant in the early stages of germination, precisely when the G1 phase is in progress. Further, the composition of these ternary complexes and their activity seem to be regulated by phytohormones during germination [26].
Since D–type cyclins participate in G1 phase, associated to Cdks and these proteins bind to PCNA, the question is if these ternary complexes are formed when PCNA is anchored to DNA. This could mean that PCNA functions as a sliding clamp so that the bound cyclin–cdk complex localizes its target proteins and phosphorylates them, which could constitute a new function of PCNA. The purpose of the present research is to determine, using the Chromatin ImmunoPrecipitation method (ChIP), if PCNA joins the cyclin–cdk complex when PCNA is associated to the chromatin.
Results
Establishing optimal ChIP conditions
As the isolation of a high number of maize embryo axes in a short time is a very heavy burden, and our initial purpose was to establish the ChIP methodology, we started using maize embryos from 21 days post anthesis kernels.
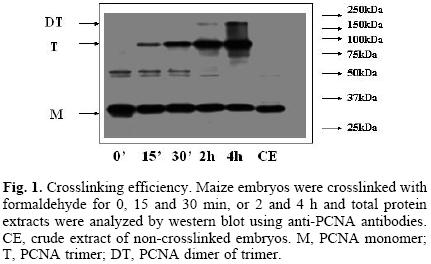
Searching for the optimal conditions for crosslinking, different times of treatment of maize embryos with 1% formaldehyde were tested (0, 15 and 30 min, 2 and 4 h). It was interesting to observe that western blots of total protein extracts, using antibodies against PCNA, showed bands of around 29, 87 and 174 kDa (Fig. 1), which appeared to correspond to the sizes of a monomer, a trimer and a dimer of a trimer of PCNA; the trimer and dimer of trimer bands increased proportionately to the increase in crosslinking time, that correlated with the consequent decrease of the monomer, 29 kDa PCNA. The appearance of the dimer of trimer band would imply the existence in plants of a PCNA novel structure, which could help explaining the great capacity of PCNA to associate to several different proteins at the same time. The nature of the bands at around 50 kDa is not known.
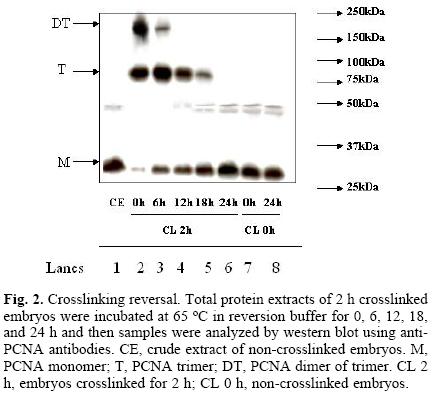
Assays in search for the optimal reversion of crosslinking time, using 2 h–crosslinked samples as starting material, were performed. Reversion times were 0, 6, 12, 18 and 24 h, at 65 °C. Figure 2 shows that the band associated to PCNA monomer increased proportionately to the incubation time whereas the intensity of the bands associated to the trimer and dimer of trimer decreased with time. The intensity of PCNA bands in non–crosslinked samples incubated for 0 and 24 h at 65 °C did not vary (Fig. 2, lanes 7 and 8), suggesting that there is no loss of protein during the processing of the samples.
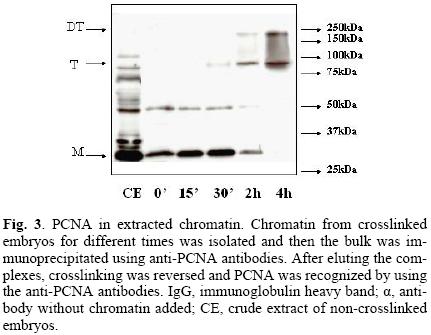
The next step was to demonstrate that PCNA was present in the chromatin pool. Figure 3 clearly shows that, after extracting chromatin, PCNA was still bound in samples crosslinked for up to two hours. That chromatin had been isolated was demonstrated by co–localizing histone H3 in chromatin extracts (data not shown).
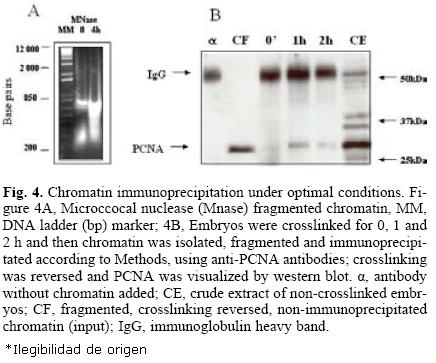
For an optimal immunoprecipitation, it is necessary to fragment the chromatin to sizes in the range of 200–600 bp DNA since longer chromatin fragments may be difficult to bring down and shorter fragments could cause the loss of both the DNA and the protein. For this purpose, micrococcal nuclease was used for different times until the desired chromatin size was obtained (Fig. 4A). Finally, optimal immunoprecipitation conditions using the anti–PCNA antibody were: optimal time of crosslinking, 1–2 h, optimal time of crosslinking reversion, 24 h, antibody dilution, 1:100 and incubation with protein A–agarose, 4 h. Figure 4B shows that under these conditions, PCNA was immunoprecipitated with crosslinking times of up to two hours; longer times appeared to make immunoprecipi–tation more difficult, probably due to the formation of more intricate or stronger nets of DNA–protein.
Association of PCNA to cell cycle protein regulators during germination
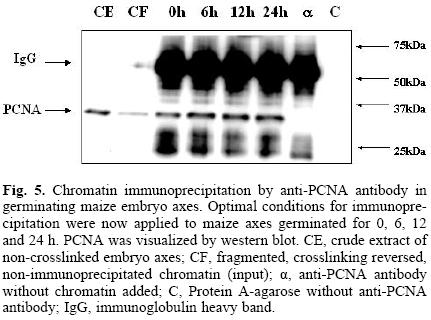
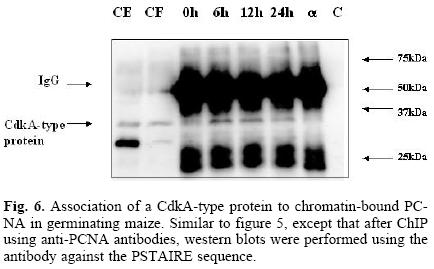
Having developed the optimal conditions for the ChIP assay, the method was now used to determine the time at which PCNA is found bound to chromatin during the germination process, and its possible association to cell cycle control proteins, namely, cyclin D4;2 and the CdkA type protein. Figure 5 shows that PCNA was bound to chromatin in maize embryo axes of dry seeds and that its amount increased as the germination process advanced. In the absence of added chromatin, or adding protein A–agarose without anti–PCNA antibody, no PCNA signal was detected. Under these conditions, it was found that PCNA bound to chromatin also associated to the CdkA–type protein (as determined by using an antibody against the PSTAIRE sequence present in all CdkA–type proteins), since this protein was present in the anti–PCNA immunoprecipitates, particularly in the early germination times and then its amount decreased by 24 h (Fig. 6).
Of the many possible existing maize D–type cyclins, Cyclin D4;2 was selected to search for its association to PCNA because there was previous evidence of this association [27] and because the antibody we have developed gives a good, clear signal by western blot. Figure 7 shows that similarly to the CdkA–type protein, cyclin D4;2 associated to PCNA when this was bound to chromatin during maize germination, although with some variation with respect to time as the cyclin, being present at time 0, diminished during the early hours of germination, to recover importantly by 24 h.

Discussion
The establishment of the methodology of chromatin immu–noprecipitation has allowed us to determine the existence of PCNA complexes with fundamental proteins for the regulation of the cell cycle in plants, a research area that has been largely neglected in the world. While this methodology was being optimized, it was found that PCNA can form more complex structures than a trimer, since the presence of a dimer of a trimer was evidenced, this is to say, two trimers that, apparently, would be sliding one very close to the other, and that therefore would be crosslinked by the formaldehyde treatment so that they would appear together in a denaturing gel. Evidence of the existence of such dimer of trimer has been found in human cells [28], although new reports corroborating these early findings have not been published. A dimer of trimer of PCNA would help explaining the capability of this protein to associate to different proteins at the same time.
PCNA has been considered as a sliding platform that is localized and bound by different proteins related to different aspects of DNA metabolism, like replication, repair or recombination, thus allowing their interaction with DNA and with other proteins to fulfill their role. One example would be the interaction of a replicative DNA polymerase with a DNA ligase during Okazaki fragment maturation, actions mediated by their binding to PCNA. However, it is not so simple to understand the association of PCNA with proteins that regulate cell cycle advancement in G1 phase because: a) there is no DNA replication going on, i.e., the S phase has not started and, b) there is no evidence that the cyclin–Cdk complex binds to the chromatin. This is the reason why it is important to determine if the cyclin–Cdk complex binds PCNA when this is associated to the chromatin. Results presented here strongly suggest that PCNA, bound to the chromatin, carries on Cyclin D4;2 and a CdkA–type protein. We do not demonstrate the existence of a ternary complex but the association of PCNA with each one of the two proteins; nonetheless, since a cyclin, or a Cdk, by itself has no apparent biological activity, it is to be expected that in fact PCNA binds to both of them, or perhaps binds one of them, lets say the cyclin and it is this cyclin that carries on the Cdk. It will be most convenient to determine which of these is the correct option. The existence of PCNA–cyclin–Cdk complexes, containing protein kinase activity, in total maize extracts has been previously described in our lab [25, 26]. The function of this ternary complex, bound to the chromatin, could be the search of phosphorylation targets, also bound to the chromatin, such as transcription factors or transcription repressors like the retinoblastoma protein (pRB or the related protein in plants, RBR) which is a well known target of G1 cyclin–kinases. This may well be the way by which, in a very specific manner, PCNA transports Cyclin D4;2 and CdkA in search of the RBR protein to phosphorylate it and thus inhibit it, therefore allowing the activation of the transcription factor E2F/DP (the S phase transcription factor) and the beginning of the S phase. Evidence of RBR phosphorylation by the Cyclin D4;2–CdkA complex has been reported previously in our lab [27].
Experimental
Plant material and imbibition
Whole maize embryos (Chalqueño variety) were hand–dissected from 21 days–post anthesis immature seeds. Maize embryo axes (Chalqueño variety) were isolated by hand, washed with sterile distilled water and were then incubated for the required time at 25 oC between sterile Whatman No. 1 paper wetted with sterile imbibition buffer containing 50 mM KCl, 10 mM MgCl2, 50 mM Tris/HCl pH 7.6, 2% sucrose and 10 μg/mL chloramphenicol.
Immunoblot assays (Western blot)
Protein (30 μg) samples were fractionated by SDS–PAGE (12%) and gels were blotted onto PVDF membranes. These were incubated with either anti–maize PCNA polyclonal antibody (dilution 1:2000), anti–maize Cyclin D4;2 polyclonal antibody (dilution 1:1000) or anti–PSTAIRE polyclonal antibody (Santa Cruz Biotechnology, dilution 1:1000) for 12 h at 4 °C, washed twice with Buffer TETN250 (100 mM Tris–HCl pH 8.0, 20 mM EDTA, 1 M NaCl, 4% Triton–X100) and three times with PBS (137 mM NaCl, 27 mM KCl and 4.3 mM sodium phosphate buffer, pH 7.2) at room temperature; subsequently, membranes were incubated for 2 h with peroxidase–conjugated anti–rabbit antibody (dilution 1:10000). Membranes were washed twice with TETN250 and 3 times with PBS and the peroxidase reaction was detected by the enhanced chemiluminescence method (ECL).
Protein crosslinking
Maize embryos or embryo axes (4 g) were crosslinked using formaldehyde at different times to form covalent bonding between proteins and proteins with DNA. Formaldehyde (1%) was applied under vacuum for 0, 15, 30 min, 1, 2 and 4 h. Reaction was stopped by adding glycine (0.15 M) under vacuum for 10 min; embryos or embryo axes were washed with distilled water to eliminate the remaining formaldehyde.
Crosslinking reversion
Reversion buffer (480 nL, 25 mM Tris–HCl pH 7.5, 10 mM EDTA, 0.5% SDS) was added to a total protein sample (20 HL) suspended in 500μL Extraction Buffer 1 (0.4 M Sucrose, 10 mM Tris–HCl pH 8.0, 10 mM MgCl2 5 mM β–mercapto–ethanol, 10 μL protease inhibitor cocktail 10X, 1 mM PMSF). This solution was incubated at 65 °C for different times (0, 6, 12, 18 and 24 h) to detect the optimal time for reversion. Proteins were precipitated using TCA (10%) and analyzed by western blot. In the case of chromatin and complexes resulting from immunoprecipitation, crosslinking reversion was at 65°C for 24 h.
DNA isolation
Once the crosslinking was reversed, proteins and DNA could be separated. For this, proteins were digested with digestion buffer (10 μL EDTA 0.5 M, 20 μL Tris–HCl pH 8.0, 1 M and 20 Hg/mL proteinase K), incubating for 1 h at 37 °C, then proteins were extracted with 500 of a chloroform:phenol:isoamyl alcohol (1:1:1) mixture and the DNA was precipitated with sodium acetate (50 μL, 3 M), glycogen (20 μg) and 2 volumes of 100% ethanol, centrifuging at 13,000 rpm. The pellet was washed with 500 μL of 70% ethanol.
Chromatin extraction
Crosslinked plant material was grounded using liquid nitrogen and fine sea sand in order to break cell walls. The resulting powder was resuspended in 20 mL of Extraction Buffer 1 to dissolve cytoplasmic proteins. This suspension was filtered and the resulting total protein suspension was centrifuged at 1800 g to precipitate organelles. The resulting pellet was resuspended in 1 mL of Extraction Buffer 2 (Extraction buffer 1 plus 0.25 M sucrose and 1% Triton X–100); the high concentration of Triton X–100 helps disrupting membranes and organelles. Proteins from broken organelles and membranes were separated by centrifugation at 20,000 g and the new pellet was resuspended in 500 uL of Extraction Buffer 3 (Extraction Buffer 2 but with 1.7 M sucrose, 2 mM MgCl2 and 0.15% Triton X–100) and this received 500 μL of Extraction Buffer 4 (Extraction Buffer 3 but with 2.5 M sucrose), added very carefully; this buffer contains a high concentration of sucrose that will help separating proteins unspecifically bound to chromatin while the chromatin was precipitated centrifuging at 124,000 g. This chromatin pellet was resuspended in 500 μL Extraction Buffer 2, washed twice with the same buffer and resuspended in 500 μL of Extraction Buffer 2.
Chromatin fragmentation
Two methods were tested: sonication and digestion by microc–cocal nuclease. Since sonication (in cycles of 40 kHz for 20 sec each) did not produce the desired chromatin size, the microc–cocal nuclease method was used. Chromatin suspended in 500 μL of Nuclease Buffer (10 mM Tris–HCl pH 8.0, 10 mM NaCl, 3 mM MgCl2, 1 mM CaCl2 1 mM PMSF) containing 10 μ/ml microccocal nuclease was incubated for 20 min at 37 °C. Reaction was stopped by adding 54 μL EGTA 50 mM, 90 μl PMSF 100 mM, 180 μL 5% SDS, 90 μL NaCl 2 M and 90 μL of a 10 X protease inhibitor cocktail. Samples (10 μL) of fragmented chromatin were resuspended in 490 μL of Reversion Buffer and DNA was separated by 1% agarose gel electrophoresis to detect the size of DNA fragments, which should be in the range of 800–200 bp.
Chromatin Immunoprecipitation
Fragmented chromatin was diluted 1:10 with dilution buffer (1.1% Triton X–100, 1.2 mM EDTA, 16.7 mM Tris–HCl pH 8.0, 167 mM NaCl) to reduce the amount of SDS of the previous buffer. Anti–maize PCNA antibody was incubated together with protein A–Agarose at 4 oC for 12 h. Then, chromatin was added and the mixture was incubated for 12 h with constant shaking at 4 oC. The mixture was washed serially with 1 mL of: Low Salinity Buffer (150 mM NaCl, 0.1% SDS, 1% Triton X–100, 2 mM EDTA, 20 mM Tris–HCl pH 8.0), High Salinity Buffer (Low Salinity Buffer but with 500 mM NaCl), LiCl Buffer (0.25 M LiCl, 1% gelpal, 1% sodium deoxycholate, 1 mM EDTA, 1 mM Tris–HCl pH 8.0) and TE Buffer (100 mM Tris–HCl pH 8.0, 10 mM EDTA). Immunoprecipitates were eluted with 250 μL of Reversion Buffer and incubated at 65 oC for 15 min. Crosslinking was reversed, proteins were precipitated and DNA separated. Proteins were analyzed by Western Blot and DNA was visualized with ethidium bromide.
Acknowledgements
Authors acknowledge receiving grants DGAPA–PAPIIT IN–201309 and CONACYT grants 42896Z, 79874 and for Formation of Young Scientists (MAM).
References
1. Miyachi, K.; Fritzler, M. J.; Tan, E. M. J Immunol. 1978, 121, 2228–2234. [ Links ]
2. Matsumoto, K.; Moriuchi, T.; Koji, T.; Nakane, P. K. EMBO J. 1987, 6, 637–642. [ Links ]
3. Bauer, G. A.; Burgers, P. M. Biochim. Biophys. Acta 1988, 951, 274–279. [ Links ]
4. Suzuka, I.; Daidoji, H.; Matsuoka, M.; Kadowaki, Y.; Takasaki, Y.; Nakane, P. K.; Moriuchi, T. Proc. Natl. Acad. Sci. USA. 1989, 86, 3189–3193. [ Links ]
5. Prelich, G.; Stillman, B. Cell 1988, 53, 117–126. [ Links ]
6. Shivji, K. K.; Kenny, M. K; Wood, R. D. Cell 1992, 69, 367-374. [ Links ]
7. Xiong, Y.; Zhang, H.; Beach, D. Cell 1992, 71, 505–514. [ Links ]
8. Tan, C. K.; Castillo, C.; So, A. G.; Downey, K. M. J. Biol. Chem. 1986, 261, 12310–12316. [ Links ]
9. Prelich, G.; Tan, C. K.; Kostura, M.; Mathews, M. B.; So, A. G.; Downey, K. M.; Stillman, B. Nature 1987, 326, 517–520. [ Links ]
10. Burgers, P. M. J. Biol. Chem. 1991, 266, 22698–22706. [ Links ]
11. Tsurimoto, T.; Stillman, B. Proc. Natl. Acad. Sci. USA. 1990, 87, 1023–1027. [ Links ]
12. Li, X.; Li, J.; Harrington, J.; Lieber, M. R.; Burgers, P. M. J. Biol. Chem. 1995, 270, 22109–22112. [ Links ]
13. Levin, D. S.; Bai, W.; Yao, N.; O'Donnell, M.; Tomkinson, A. E. Proc. Natl. Acad. Sci. USA. 1997, 94, 12863–12868. [ Links ]
14. Umar, A.; Buermeyer, A. B.; Simon, J. A.; Thomas, D. C.; Clark, A. B.; Liskay, R. M.; Kunkel, T. A. Cell 1996, 87, 65–73. [ Links ]
15. Gary, R.; Ludwig, D. L.; Cornelius, H. L.; MacInnes, M. A.; Park, M.S. J. Biol. Chem. 1997, 72, 24522–24529. [ Links ]
16. Chuang, L. S.; Ian, H. I.; Koh, T. W.; Ng, H. H.; Xu, G.; Li, B. F. Science 1997, 277, 1996–2000. [ Links ]
17. Kelman, Z. Oncogene 1997, 14, 629–640. [ Links ]
18. Waga, S.; Hannon, G. J.; Beach, D.; Stillman, B. Nature 1994, 369, 574–578. [ Links ]
19. Koundriouckoff, S.; Jonsson, Z. O.; Hasan, S.; de Jong, R. N.; van der Vliet, P. C.; Hottiger, M. O.; Hubscher, U. J. Biol. Chem. 2000, 275, 22882–22887. [ Links ]
20. Sherr, C. J. Cell 1994, 79, 551–555. [ Links ]
21. Pagano, M.; Theodoras, A. M.; Tam, S. W.; Draetta, G. F. Genes Devel. 1994, 8, 1627–1639. [ Links ]
22. Tsurimoto, T. B. B. Acta 1998, 1443, 23–39. [ Links ]
23. Nakagami, H.; Sekine, M.; Murakami, H.; Shinmyo, A. Plant J. 1999, 18, 243–252. [ Links ]
24. Gutierrez, C.; Ramirez–Parra, E.; Castellano, M. M.; del Pozo J. C. Curr. Opin. Plant Biol. 2002, 5, 480–486. [ Links ]
25. Sánchez, M. P.; Torres, A.; Boniotti, M. B.; Gutiérrez, C.; Vázquez–Ramos J. M. Plant Mol. Biol. 2002, 50, 167–175. [ Links ]
26. Sánchez, M. P.; Gurusinghe, S. H.; Bradford, K. J.; Vázquez–Ramos, J. M. J. Exp. Bot. 2004, 56, 515–523. [ Links ]
27. Lara–Núñez, A.; de Jesús, N.; Vázquez–Ramos, J. M. Physiol. Plant. 2008, 132, 79–88. [ Links ]
28. Naryzhny, S. N.; Zhao, H.; Lee, H. Cell 2005, 280, 13888–13894. [ Links ]
Dedicated to Dr. Estela Sánchez de Jiménez for her invaluable contributions to plant biochemistry.
1 Both authors contributed equally to this work.

