











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
Amyloid transthyretin (ATTR) amyloidosis is a genetic disease resulting from misfolded TTR protein deposition in multiple organs. Its prevalence has been described worldwide1. Cardiomyopathy and sensory-motor and autonomic neuropathy are the hallmark manifestations, leading to progressive morbidity and a fatal outcome if they remain unrecognized and untreated. Besides, there is high variability among symptoms even in individuals of the same family, which could make the diagnosis challenging2,3. Therefore, a complete neurological and autonomic assessment will be essential for these patients' early diagnosis.
Introduction to amyloid and amyloidosis
Amyloid is a sub-microscopic fibrillary structure composed of low-molecular-weight subunits of various serum proteins with a mean diameter of 10 nm. It is characterized by an antiparallel B-pleated sheet structure, binding to Congo red, leading to apple green birefringence under polarized light. Amyloidosis is a generic term that refers to the disease caused by the deposition of this material on the extracellular surface of organs and tissues. It may be hereditary, secondary to a spectrum of inflammatory conditions, or a member of plasma cell neoplasm4,5.
At present, 18 proteins appearing as systemic amyloidosis and 22 as localized forms have been identified. In terms of nomenclature, the amyloid fibril protein is designed as protein A and followed by a suffix, an abbreviated form of the parent or precursor protein name. Relevant systemic amyloid types are shown in figure 1. The most common forms are those amyloid fibrils derived from immunoglobulin light chains (AL), amyloid transthyretin (ATTR), and serum amyloid (AA)5,6.
Hereditary amyloidosis
TTR is a tetrameric protein composed of 127 amino acids synthesized in the liver. It is a transporter of thyroxin (T4) and retinol (Vitamin A)-binding protein. The production also occurs in the brain's choroid plexus and the eye's retinal pigment epithelium2. TTR gene alterations cause dissociation of the tetramer into monomers, forming amyloid deposits in several organs, producing organ dysfunction, and resulting in ATTR amyloidosis. There are two major forms of ATTR amyloidosis: hereditary ATTR amyloidosis named ATTRv (v for variant), wild-type ATTR amyloidosis (ATTRwt), and acquired ATTR amyloidosis in domino liver transplantation3.
Although there are other forms of hereditary amyloidosis, ATTR amyloidosis is the most common. Less common hereditary forms include apolipoprotein I amyloidosis (AApoAI), cystatin C amyloidosis, and fibrinogen alpha amyloidosis, among others. There are nearly 136 genetic variants for ATTR amyloidosis and 60 for the other types6 (Fig. 1).
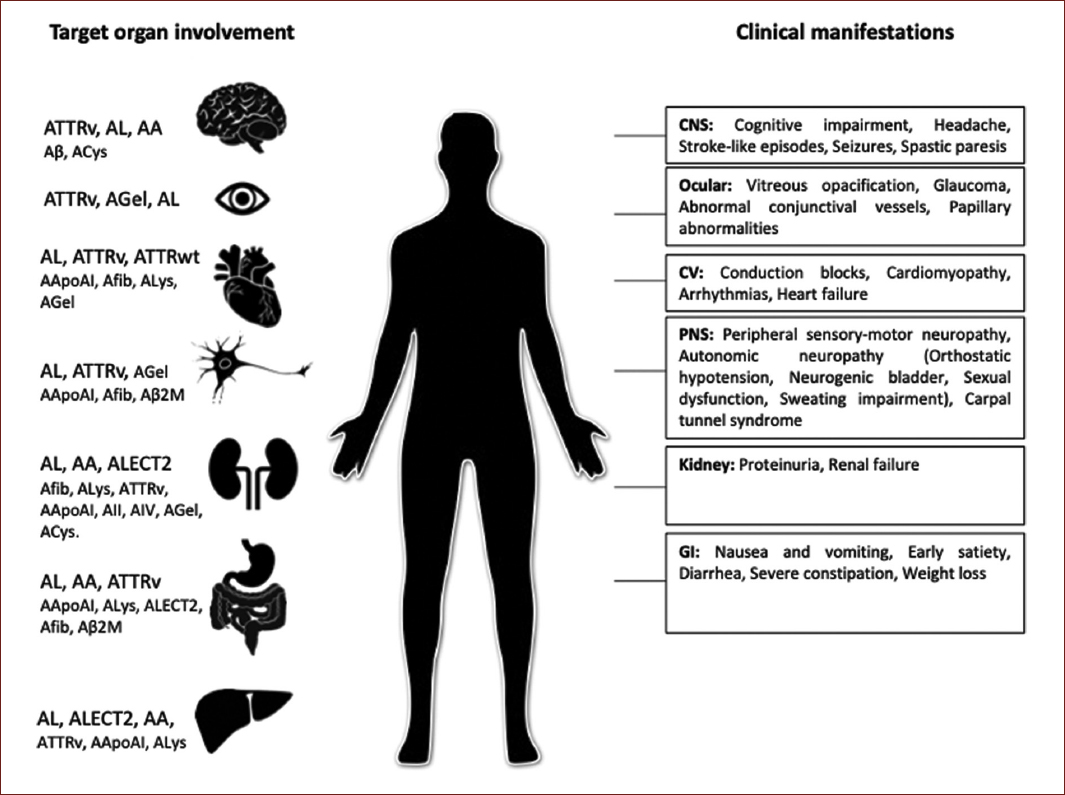
Figure 1 Systemic amyloidosis types by target organ involvement and specific clinical features. ATTRv: transthyretin variants; AL: immunoglobulin light chain; AA: (Apo) serum amyloid A; Aβ: Aβ protein precursor; ACys: cystatin C; AGel: gelsolin; ATTRwt: transthyretin wild type; AApoAI: apolipoprotein AI; Afib: fibrinogen; ALys: lysozyme; Aβ2M: β-2 macroglobulin; AApoAII: apolipoprotein AII; AApoAIV: apolipoprotein A IV; ALECT2: leukocyte chemotactic factor-2; CNS: central nervous system; CV: cardiovascular; PNS: peripheral nervous system; GI: gastrointestinal.
Genetics and demographics of ATTRv amyloidosis
ATTRv amyloidosis was first described in a fisherman's town in Portugal in 1952. In the following two decades, cases were reported in other parts of the world, particularly in Japan and Sweden. Today is been diagnosed worldwide1.
The TTR gene is located in chromosome region 18q12.1. Point mutations in this gene cause ATTRv amyloidosis which has autosomal dominant inheritance with variable phenotypic expressions and penetrance1,2,7; some evidence states that affected women transmit higher disease penetrance. Genetic anticipation has been observed in endemic regions. More than 130 variants were described, and their prevalence depends on geographic region1. Val30Met is the most prevalent ATTRv amyloidosis mutation in the world, with the largest cluster of individuals located in North Portugal1,7. In Mexico, the most prevalent mutations are Ser50Arg, Gly47Ala, and Ser52Pro, which were found mainly in Guerrero, Morelos, and Guanajuato7,8.
The prevalence in Europe is estimated at 1/100,000 individuals1 but is highly variable as in endemic areas such as North Portugal is up to 151/100,000 individuals9 and in Mexico is 0.89/100,000 individuals7.
The age at onset of disease-related symptoms varies between the second and ninth decades of life, with significant disparity across different populations. Portugal and Japanese foci of patients have traditionally been described as early-onset (mean age, 33 years), meanwhile Swedish as late onset (56 years)1,9. Mexican population reported a mean age at onset of 35 years7. Patients with early onset may present high penetrance10. The male-to-female ratio is of 1:11,2,7,9. The mean duration of the disease is approximately 10 years, ranging from 6 to 12 years. Nevertheless, the Mexican series reported between 1 and 4 years since disease diagnosis, as 10% of their patients were diagnosed in advanced stages7. The cause of death is frequently either sudden death or cardiac failure1,10.
Clinical spectrum
As mentioned before, ATTRv amyloidosis differs considerably between patients, depending on the mutation, age at onset, and if it is an endemic zone2,3,10; it could vary even within the same family. In addition, epigenetic and environmental factors can play a role11.
Genotype and phenotype correlations
Genotype and population origin are essential determinants of symptoms. Some mutations will induce cardiomyopathy as the predominant feature (e.g., Val122Ile, Ile68Leu, Thr60Ala, and Leu11Met), while others are associated primarily with neuropathy (e.g., Val30Met), but both manifestations can be present in different proportions1,10,12 (Fig. 2).
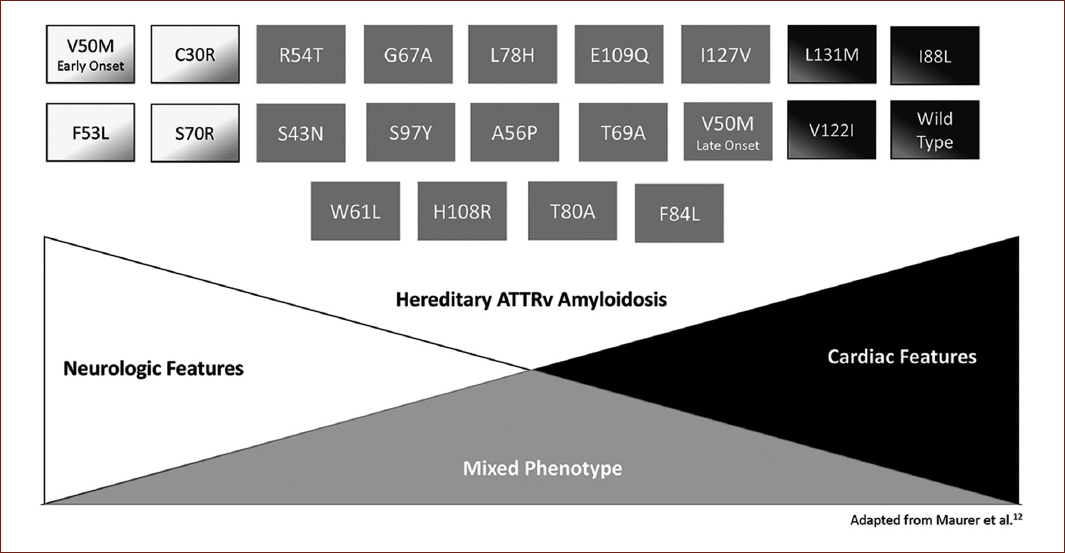
The endemicity of a region will be another determinant factor, as endemic areas show an earlier onset, higher penetrance, more severe manifestations, and a higher index of detection7,10,13.
However, the genotype-phenotype correlation is more complex as the clinical presentations can also be influenced by other factors such as non-coding variations of the TTR gene and the expression in tissues13. The most striking example of inter-population diversity is the Val30Met mutation. In Val30Met Portuguese families, the disease shows early onset, substantial severity, and high penetrance, whereas Val30Met in Swedish patients has late onset and low penetrance10,13.
In Mexico, the mutations described Ser50Arg, Gly47Ala, Ser52Pro, and V122I/Y116H behave similarly to endemic populations as the onset of clinical manifestations is very early (35 years), the picture is more aggressive in males than females, and the manifestations appear earlier in younger generations7.
Neurological manifestations
PERIPHERAL NEUROPATHY
In both endemic and non-endemic areas, a symmetric axonal length-dependent sensory-motor and autonomic polyneuropathy is a hallmark manifestation but not always the debut10.
Amyloidosis typically affects first distal small myelinated and unmyelinated nerve fibers altering pain and temperature sensation, which may manifest as paresthesia, dysesthesia, allodynia, or hyperalgesia in the feet. In addition, the clinical examination may detect impaired thermal sensitivity in the feet with decreased pinprick sensation1,11.
Axonal degeneration then progresses relentlessly in a distal to proximal pattern reaching upper limbs, usually 4-5 years after. Within a few years, larger myelinated sensory and motor nerves become affected, and impairment of light touch, vibrations, and position sensation and motor deficit appears in the distal lower limbs. Thus, they start with gait disturbances characterized by sensory ataxia and weakness that progress until they become wheelchair dependent or bedridden. Motor deficits also follow length-dependent progression1,10,11,14.
Wang et al. described five patterns of polyneuropathy in patients with amyloidosis. The most frequent in his series was polyneuropathy with pain and generalized autonomic failure (GAF), followed by polyneuropathy and GAF without pain, and somatic small-fiber neuropathy and GAF. The GAF-only pattern was less common than those above15.
The peripheral autonomic nervous system is susceptible to amyloid infiltration and injury, whereas the central nervous system remains almost unaffected; the reason has not been elucidated16.
Autonomic neuropathy
One significant finding that is present more often in ATTRv amyloidosis than in other types of amyloidosis is autonomic neuropathy, which has been only reported in 10-14% of patients with AL amyloidosis and is considered exceedingly rare in the AA form. Therefore, autonomic symptoms are vital components of ATTRv amyloidosis, contributing enormously to the burden of the disease in 73% of patients10,17.
As mentioned before, unmyelinated and small myelinated nerves are affected before large fibers. Thus, autonomic dysfunction appears very early, even before somatic sensory symptoms manifest. In addition, more extensive amyloid deposits have been found in the spinal ganglion and the posterior root of the spine than in the anterior root. There is also peripheral catecholamine depletion16,17.
Autonomic dysfunction affects the cardiovascular, gastrointestinal, urinary, ocular, and sudomotor systems, among others15-18.
CARDIOVASCULAR DISTURBANCES
The most dangerous autonomic manifestation is cardiovascular autonomic neuropathy due to amyloid infiltration of nerves controlling peripheral blood flow and blood pressure. The primary manifestation is neurogenic orthostatic hypotension, in which we can see a fall in blood pressure >20/10 mmHg while standing with a lack of pulse response. Moreover, amyloid deposits in the heart's conductive system provoke cardiac arrhythmias, which in association with diminished sympathetic responses and parasympathetic denervation supersensitive, can lead to sudden death16-18.
Another objective marker of autonomic dysfunction in these patients is the low heart rate variability (HRV) which depicts parasympathetic failure and can be affected very early in the disease16,18.
GASTROINTESTINAL SYMPTOMS
Gastrointestinal manifestations are common in ATTRv amyloidosis, and patients invariably develop severe disturbances during the disease10,16,17. These manifestations result from a loss in the inhibitory and increase in the excitatory enteric neurons, which may result mainly in gastroparesis and dysmotility17.
The initial symptoms, severity, and evolution depend on the genetic variant, but classically, patients have pronounced constipation with bouts of diarrhea. In the end stages of the disease, the patients suffer from continuous diarrhea and fecal incontinence, often accompanied by postprandial vomiting; they become unable to retain a sufficient food intake.
After the onset of gastrointestinal dysfunction, continuous weight loss is observed, leading patients to severe malnutrition, which correlates with survival16.
GENITOURINARY FUNCTION
Early damage of the sacral parasympathetic fibers contributes to genitourinary dysfunction, starting from increased urine retention to nycturia, incomplete bladder emptying, and urgency up to overflow incontinence due to the progressive involvement of motor sympathetic and somatic nerves. Urinary tract infections contribute to the morbidity17,18. Erectile dysfunction is another early manifestation in males16-18. Kidney involvement is rare unless is consequence of chronic urine stasis or cardiorenal syndrome10,16.
PUPILLOMOTOR AND SUDOMOTOR FUNCTIONS
Amyloid deposits also can be found in perivascular areas, in and around the nerves, and stoma of the extra bulbar tissue, ciliary body, choroid, retina, and the vitreous body. Vitreous opacity is a typical manifestation of the disease in certain families16. Pupillary abnormalities are induced by autonomic dysfunction due to deposits and the destruction of the nerves. The sympathetic predominance in pupil control decreases its diameter at rest. Scalloped pupils, defined as bilateral, irregular pupillary margins and fringed edges, are a unique sign17. Dry eye is another early autonomic symptom1.
Patients have inappropriately cold hands and feet with discoloration, suggesting that the vasoregulation of the peripheral vessels is impaired. These vasomotor changes, in addition to pain insensitivity, result in chronic ulceration and mutilation of the acral extremities17.
Carpal tunnel syndrome
Carpal tunnel syndrome can be present in up to two-thirds of patients with ATTRv amyloidosis neuropathy and can precede the diagnosis by several years. This finding is caused by compression of the median nerve due to amyloid deposition in the flexor retinaculum19. It should be noted that many patients are erroneously diagnosed with simple carpal tunnel syndrome, progressive symptoms, or lack of improvement after carpal tunnel release surgery and the coexistence with other systemic symptoms should raise the suspicion of ATTRv amyloidosis1,19.
CNS MANIFESTATIONS
The acknowledgment of the leptomeningeal form of ATTRv amyloidosis is recent. In this case, it is believed that the source of variant TTR is not the liver but the choroid plexus1. Moreover, several studies show CNS symptoms arise as a typical late complication in patients with the V30M mutation after at least 14 years of symptomatic peripheral nerve disease. Manifestations in these cases involve transient focal neurologic episodes, hemorrhagic and ischemic stroke, cognitive decline, and cranial nerve dysfunction1,20.
Cardiac manifestations
In both hereditary and non-hereditary TTR amyloidosis, as in AL amyloidosis, amyloid can infiltrate any or all of the cardiovascular structures, including the conduction system, the atrial and ventricular myocardium, valvular tissue, and the coronary and large arteries. Myocardial infiltration progressively increases the thickness of the left and right ventricular walls and interventricular septum. Cardiac amyloidosis is generally considered cardiomyopathy with a hypertrophic phenotype and restrictive pathophysiology. The left ventricular ejection fraction is normal or only mildly reduced. The involvement of cardiac valves leads to the formation of nodules or diffuse thickening of the leaflets, accompanied by variable degrees of valvular regurgitation. Regarding the conduction system, bundle branch block, atrioventricular, and sinoatrial blocks are the most frequent findings1,12,21.
Patients with ATTR cardiomyopathy (ATTR-CM) commonly present with dyspnea, fatigue, and edema but with preserved ejection fraction. On the echocardiogram, moderate-to-severe left ventricular thickening (wall thickening > 14 mm) should trigger consideration of ATTR-CM, especially if there is discordance between wall thickness and QRS voltage on ECG. Elevated troponins or N-terminal pro-brain natriuretic peptide (NT-proBNP) levels that are out of proportion to the clinical context and evidence of the right-sided heart failure with intolerance to ACE inhibitors or beta-blockers are some other pitfalls that should raise suspicion12,21.
Skin manifestations
In ATTRv amyloidosis, cutaneous involvement is not as frequent as in AL. The most common findings are xerosis, seborrheic dermatitis, acne, neurotrophic ulcers, and periorbital purpura. These symptoms are usually present only in later stages22.
Red flags and differential diagnosis
Patients with ATTRv amyloidosis present with non-specific symptoms that are frequently attributed to more common disorders, predominantly in the initial stages; this creates a challenge for a correct and early diagnosis1,10.
Characteristic features that can be useful in identifying this disease are heritability and general multi-symptom involvement. Therefore, it is vital to obtain a complete clinical history with details of symptoms of systemic disease and the family history. The presence of a progressive and symmetrical length-dependent sensory-motor polyneuropathy and at least one of the following may raise suspicion: family history of neuropathy, early autonomic dysfunction regarding cardiovascular, genitourinary, or gastrointestinal features, cardiac involvement, inexplicable weight loss, refractory carpal tunnel, or vitreous opacity10,11.
The most frequent combination of early symptoms are predominantly small-fiber peripheral polyneuropathy with autonomic features, such as erectile dysfunction, gastrointestinal dysfunction with weight loss, and peripheral polyneuropathy with cardiac manifestations11. Length-dependent pattern of symmetric sensory-motor and autonomic polyneuropathy in ATTRv amyloidosis is not unique. The most common neuropathic misdiagnosis for the sporadic form is chronic inflammatory demyelinating polyneuropathy (CIDP). It has been reported in different series that up to 20% of patients were initially misdiagnosed with CIDP. Although CIDP is generally characterized by demyelinating nerve damage, once an extensive axonal injury is present, electrophysiological characteristics of ATTRv amyloidosis can resemble those of CIDP due to secondary demyelination. On the other hand, protein levels in cerebrospinal fluid can also be elevated in both diseases, but more markedly in CIDP patients. Although the poor response to immunomodulatory treatment in patients is first classified as CIDP, it is another feature that should make the clinician consider other possibilities10,11.
Another common differential diagnosis of the symmetrical axonal sensory-motor and autonomic polyneuropathy is diabetic polyneuropathy, which also affects small somatic fibers in a length-dependent way first. Nevertheless, the long evolution of non-control diabetes should be essential with the autonomic dysfunction in the later stages of the disease. The severe weight loss combined with the initial sensory polyneuropathy could make us consider paraneoplastic neuropathy first. However, the prominent ataxia in the early stages, the usually non-length dependent sensory loss, and a negative PET and negative anti-onconeural antibody panel should make us consider other possibilities10.
Conversely, monoclonal gammopathy in elderly patients or false immunolabeling of amyloid deposits could lead to misdiagnosed AL amyloidosis instead of ATTRv and result in inappropriate chemotherapy treatment. Besides, a substantial proportion of patients with ATTRv also have MGUS. This situation can be avoided by careful typifying of the amyloid precursor protein11,23,24.
Wild-type amyloidosis
Several amyloidosis are associated with aging, including systemic and localized forms. The development of these amyloidosis may be related to the age-related reduced effectiveness of the protein folding quality control systems. With aging, the TTR tetramer becomes less stable, releasing misfolded intermediates that form amyloid deposits, mainly in the heart24.
ATTR derived from the wild-type protein (ATTRwt), formerly called "senile systemic amyloidosis," is likely the most common type of cardiac amyloidosis. The median age at diagnosis is 75 years, and approximately 90% of patients are men - two-thirds of affected individuals present with heart failure, atrial flutter or fibrillation, and other conduction abnormalities. Furthermore, carpal tunnel syndrome, often bilateral lumbar spinal stenosis and bicep tendon rupture, is present in 30-50% of patients. Other organs, albeit less symptomatic, can be involved and may include peripheral nerve, lungs, gastrointestinal tract, gallbladder, prostate, and bladder. The median survival of ATTRwt is 3.6 years23,24.
Diagnosis
Neurological and autonomic evaluation
The neurologic examination in a patient with suspicion of ATTRv amyloidosis should encompass the complete neuropathy assessment, including nerve conduction velocities with electromyographic testing to evaluate large fibers, quantitative sensory testing for small somatic fibers, and proper autonomic testing1.
Regarding cardiovascular autonomic evaluation, HRV is a good marker of parasympathetic dysfunction and is proportional to the severity of the neurologic disability. HRV can be assessed with the heart rate deep breathing test, the Valsalva index, spectral analysis, and time-domain indices by 24 h Holter17,18,25.
The cardiovascular sympathetic system could be evaluated with the Valsalva phases in a blood pressure beat-to-beat monitoring and with the tilt test. Further, an active orthostatic test could be done at the bedside, asking the patient to lie down for 5 min and then stand up while recording the brachial systolic and diastolic blood pressure and heart rate in both positions17,18.
Other useful method is the cardiac 123I-meta-iodobenzylguanidine scintigraphy, a sensitive method for the detection of postganglionic sympathetic denervation of the heart, which will show no uptake in patients with ATTRv amyloidosis17,18.
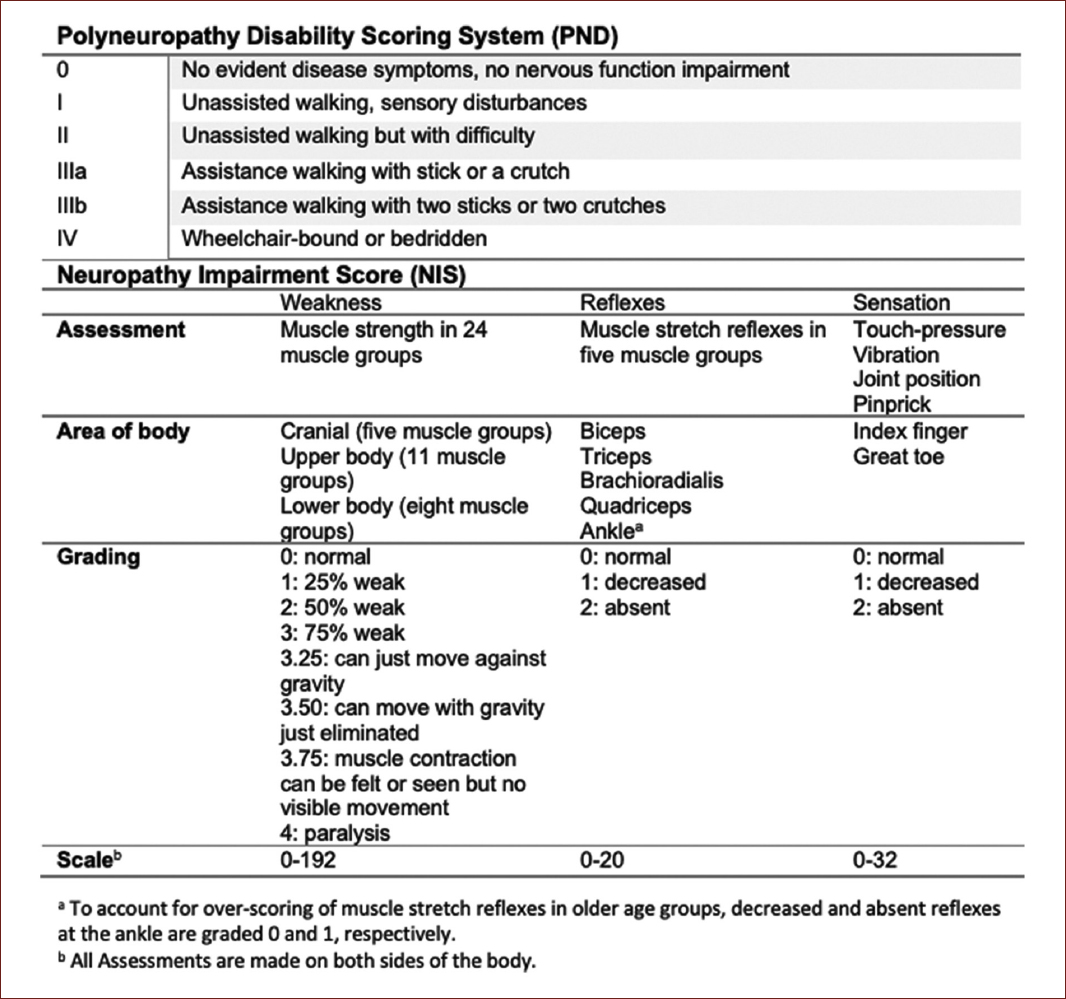
Some scales have been developed to measure the extent of disability of the neuropathic burden in these patients. The firsts used were the familial amyloidotic polyneuropathy staging system and the polyneuropathy disability scoring system. However, these scores involve a greater separation of disease stages, making them insensitive to tracking disease progression over shorter periods and impractical for use as primary outcome measures in clinical trials. Therefore, the neuropathy impairment score (NIS) and its variations the NID-lower limb (NIS-LL) and the NIS+7 are more sensitive instruments, as this last scale includes nerve conduction studies and quantitative sensory and autonomic endpoints (Fig. 3)26.
Cardiac evaluation
All patients with suspicion of ATTRv amyloidosis should be referred to an amyloid expert cardiologist. Cardiac evaluation should include ECG, echocardiography, BNP, and troponin measurement, and if possible, the cardiac resonance can be useful in tissue characterization and distinguishing amyloid from non-amyloid wall-thickening disorders. However, myocardial scintigraphy with bone avid tracers as 99mTechnetium-pyrophosphate (99mTc-PYP) and 99mTechnetium-3,3-diphosphono-1,2-propanodicarboxylic acid (99mTc-DPD), has a higher sensitivity and specificity for cardiac ATTR amyloid deposits12,27. The 99mTc-DPD scintigraphy has demonstrated greater binding avidity since the early stages27. Nowadays, with these new image techniques, cardiac biopsy has become the last resort21.
Biopsy
Deposition of amyloid in the tissue can be demonstrated by Congo red staining of biopsy specimens. Amyloid deposits show a characteristic green birefringence under polarized light. Fat tissue has been the most accessible for diagnosis, but its yield is amyloid type dependent, ranging from 15% to 80%. Other typical tissue sources are the gastrointestinal mucosa with 85% sensitivity, minor salivary gland, kidney, sural nerve, and bone marrow1,10,24.
A negative biopsy does not exclude the existence of amyloid deposits, given the high rate of false-negative results. In the same line, a positive biopsy is seldom needed to begin treatment in clinically symptomatic carriers of the mutation, but may be still a legal/regulatory requirement in some countries28.
The amyloid deposits in all forms of amyloidosis are histologically similar. Therefore, amyloid typing is crucial; the most reliable method is mass spectrometry (MS) proteomic-based analysis, which directly identifies the protein subunit in the deposit (AApoAIV, ALECT2, and AA). This technique is expensive and not widely available. Although less sensitive and specific than MS, immunohistochemistry, immunofluorescence, and immunoelectron microscopy are acceptable alternative methods23,24.
Genetic diagnosis
More than 130 mutations in the TTR gene can occur, most of which are pathogenic. Thus, DNA sequencing is usually required to know the site and kind of amino acid substitution in several disease-related TTR gene mutations, which will play an essential role in defining the disease course and prognosis1,10.
Treatment
Disease-modifying treatments
Disease-modifying treatments have only been tested in clinically symptomatic ATTRv patients; pre-symptomatic treatment of gene carriers is not an accepted indication at this time. These patients should be followed up annually, and if they show any early sign of sensory-motor neuropathy, carpal tunnel, autonomic symptoms, unexplained weight loss, cardiac hypertrophy, arrhythmias, cardiomyopathy, renal abnormalities, or ophthalmologic manifestations, they must initiate treatment. Early biomarkers of the disease are under investigation, particularly amyloid deposits in skin biopsies, decreased intraepidermal nerve fibers, and serum neurofilament light chain29. In patients with cardiac involvement, Tc99 or PYP PET scan is excellent tools that aid in the diagnosis of ATTR infiltration in the heart30.
LIVER TRANSPLANT
Available therapeutic approaches comprise reducing and stabilizing circulating TTR to prevent the dissociation of tetramers into monomers. The liver is the primary source of circulating TTR, so orthotopic liver transplantation became in 1990, one of the leading options for treatment. Its use has declined since the availability of disease modifying treatments; however, it is still an option in countries where other modifying treatments are not affordable. The long-term efficacy from the survival point of view has been proven and will depend on the mutation, the patient's age, and the disease stage. In some cases, the progression of cardiomyopathy and neuropathy resulting from wild-type TTR deposition may occur even after, particularly in late-onset male patients. Overall patient survival rate at 10 years is 74% in Val30Met patients versus 44% in non-Val30Met1-3. Combined liver-heart/kidney has been a life-saving approach for patients with advanced disease1,3.
TTR STABILIZERS
TTR stabilizers aimed to stabilize the native quaternary structure of TTR tetramers using small molecules that bind to thyroxin-binding sites to avoid protein misfolding3.
In the early 2010s, tafamidis and diflunisal, two oral TTR stabilizers, demonstrated by randomized controlled trials their efficacy for ameliorating the progression of neuropathy31,32.
Tafamidis has proven its long-term efficacy and safety for up to 6 years33 due to ATTRv and ATTRwt amyloidosis34. Compared with placebo, it has decreased 30% mortality for all causes and admissions due to cardiovascular complications by 32%. This medication is also approved for ATTRv Stage 1 polyneuropathy; the results have been variable in patients with Val30Met ATTRv in advanced stages and non-Val30Met patients at any stage33.
Diflunisal, a nonsteroidal anti-inflammatory drug, demonstrated its efficacy by decreasing neuropathy progression measured by the NIS+7 scale, but chronic use can cause renal disease, and it is not approved for use3,32.
TTR GENE-SILENCING DRUGS
TTR seems to exert harmful effects even when fibrillar structures recognized as amyloid fibrils are not formed; it can induce microangiopathy, which plays a role in organ damage. Thus, the development of gene-silencing therapeutics, including small interfering RNA (siRNA) and antisense oligonucleotide (ASO), aims to eliminate circulating TTR3.
Inotersen is a second-generation subcutaneous ASO that demonstrated significantly better primary endpoints represented by neuropathy impairment and quality of life scores for 8 months of use with a TTR reduction of 79%. Nevertheless, glomerulonephritis and severe thrombocytopenia were reported as severe adverse events; hence, close monitoring of renal function and platelet count is required in every patient receiving this drug35.
Patisiran is an intravenous siRNA formulated in a lipid nanoparticle delivered to the liver; it reduces mutated TTR production interfering with mRNA by the RNA-induced silencing complex. The response to treatment was observed broadly with decreased TTR production in 84% at 18 months and an improvement in almost every endpoint, including autonomic symptoms, compared to placebo. This medication requires pre-medication; the adverse events most frequently reported were peripheral edema and infusion-related reactions36.
Vutrisiran is a subcutaneous formulation that utilizes the same siRNA approach as patisiran to target variant and wild-type TTR synthesis. This medication significantly improved multiple disease-relevant outcomes for ATTR amyloidosis versus external placebo, with an acceptable safety profile. Furthermore, TTR reduction was non-inferior to within-study patisiran and resulted in a reduced peak-to-trough fluctuation37. Consequently, the FDA approved Vutrisiran in June 2022 to treat the polyneuropathy of ATTRv amyloidosis in adults38.
Symptomatic treatment
Once patients have developed symptoms, treatment should also focus on alleviating them using specific medications for each complication, that is, fludrocortisone or midodrine for orthostatic hypotension, pacemaker implantation, and antiarrhythmic drugs for arrhythmias and prokinetics for gastroparesis, among others1,3,17. The use of diuretics, antihypertensive medications like angiotensin-converting enzyme inhibitors, or inhibitors of phosphodiesterase type 5 for erectile dysfunction should be used with extreme caution to avoid increasing orthostatic hypotension.
The goal will always be to have a multidisciplinary approach and establish a short- and long-term strategy for treating these patients.
Conclusions
ATTRv amyloidosis is a challenging genetic disease with neurological and systemic manifestations frequently misdiagnosed. As a result, it could severely affect the quality of life resulting in fatal outcomes in a relatively short time. As leading referring physicians, neurologists play a key role in changing the natural course of this illness by promoting early recognition and treatment. Therefore, future guidelines should focus on raising awareness of the diagnostic pitfalls and current disease-modifying treatments of ATTRv amyloidosis.

