











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Nonalcoholic fatty liver disease (NAFLD) is the most prevalent chronic liver disease and is strongly associated with obesity and the attendant metabolic syndrome.1 NAFLD encompasses a broad spectrum of liver disorders, ranging from simple steatosis to its more severe form, nonalcoholic steatohepatitis (NASH), which progresses to cirrhosis or hepatocellular carcinoma in 20-25% of patients.1 Oxidative stress is believed to be an important contributor to the pathogenesis of NASH.2 Excess hepatic iron can promote oxidative stress via Fenton’s reaction and is proposed to be a cofactor in the development of NASH.2 We have previously shown that the presence of hepatic iron and the occurrence of HFE mutations such as C282Y, is associated with more advanced NASH suggesting a role for iron in exacerbating NAFL to NASH.3-5 Further, we have also shown a strong relationship between hepatic iron deposition pattern and histologic severity of NAFLD wherein iron deposition in hepatic reticuloendothelial system (RES) cells is associated with higher NAFLD activity score (NAS) and advanced fibrosis.5 Thus, the involvement of iron in the pathogenesis of NASH underscores the need to better understand the iron homeostasis pathway, with emphasis on the regulatory role of hepcidin, a key iron homeostasis protein.6
Hepcidin, produced primarily by hepatocytes, regulates intestinal iron absorption, macrophage iron recycling andmobilization of iron from hepatic stores by binding and internalizing the iron transporter, ferroportin.6 Hepcidin expression is enhanced by increased iron stores via hemojuvelin (HJV), bone morphogenetic protein (BMP6) and Sma and Mad related proteins (SMAD) via HJV/ BMP/SMAD pathway7 and during inflammation via induction of Janus kinase/Signal transducer and activator of transcription (JAK2/STAT3) pathway,8,9 conditions commonly seen in NAFLD.10 Hepcidin is inhibited by anemia, hipoxia and augmented erythropoiesis via the transcription factor C/EBPa and transmembrane serine protease 6 (TMPRSS6).9,11,12-14 Hepcidin regulatory mechanisms have been investigated in animal models and only a few studies have investigated the role of hepcidin in human NAFLD patients, 10,15-18 none have examined the regulatory mechanisms of hepcidin expression in patients with NAFLD.
The aim of this study was to examine the expression profiles of iron regulatory genes, and inflammatory and oxidative and other stress-response pathways in patients with NAFLD, with a goal to identify differentially expressed genes in patients who have NASH, and in NAFLD patients with and without hepatic iron overload. Total RNA from liver tissues of patients with NAFLD were analyzed to evaluate expression profiles of candidate genes in the disease states; NAFL vs. NASH, and based on presence or absence of observable iron deposition in liver tissue of NAFLD patients.
Material and methods
Subjects
Patients were enrolled from University of Washington Medical Center and Virginia Mason Medical center clinics in Seattle, WA in the study. The study was approved by the human subjects institutional review board and written informed consents were obtained from all subjects. Inclusion criteria were: minimal alcohol use (< 20 g/day) reported by the patients and appropriate exclusion of other liver diseases including viral hepatitis, autoimmune hepatitis, drug-induced liver disease, primary biliary cirrhosis, Wilson’s disease, and α1-antitrypsin deficiency. Hematoxylin and eosin staining along with Prussian Blue stain specific for iron were used in the grading of the NAS score. All patients met the criteria for NAFLD as defined by Brunt, et al.19
Baseline laboratory studies
Clinical data included body mass index (BMI), presence of diabetes mellitus, serum aminotransferase levels Aspartate transaminase (AST) and Alanine transaminase (ALT), triglycerides, cholesterol, glucose, iron, ferritin and transferrin iron saturation. Liver tissue was obtained by percutaneous needle biopsy and samples were snap frozen in liquid nitrogen, stored at -70 ºC for subsequent RNA extraction and gene expression analysis.
RNA preparation and cDNA synthesis
Total RNA was isolated from liver tissue using RNeasy Mini kit (QIAGEN GmbH, Germany). RNA quality was determined using RNA 6000 Nano Chip Kit and Bioanalyzer 2100 (Agilent Technologies). Samples with a RNA Integrity Number (RIN) value of 4.0 or greater were included for gene expression studies. The cDNA first-strand synthesis was performed using Superscript First-Strand Synthesis System (Invitrogen) and oligo (dT) primers with 1 μg RNA in 20 μL reaction volume.
Quantitative real time PCR: Quantitative real time-PCR using SYBR GreenER qPCR super mix (Invitrogen) was performed on the ABI 7900 HT real time PCR machine. Primers were designed to span intronic sequences to avoid amplification of any contaminant genomic DNA. Specific primers were used for RT PCR amplification of HAMP (hepcidin antimicrobial peptide), FPN1 (ferroportin), CREBH (cAMP responsive element-binding protein, hepatocyte specific), IL-6, IL-6R, IL-1β, TNFα, NFκB, BMP6 (bone morphogenetic protein), TMPRSS6, SOCS3 (suppressor of cytokine signaling 3), HO-1 (hemeoxygense 1), HIF1α (hypoxia inducible factor-1α) and STAT3 genes and are tabulated (Table 1) and HJV primers obtained from Applied Biosystems were used. 10 μL of SYBR GreenER qPCR super mix (Invitrogen), 1 μM of (forward and reverse) primers and 25 ng of the cDNA template were mixed in a 20 μL reaction volume. Fold change in gene expression was determined by normalization to glyceraldehyde phosphate dehydrogenase (GAPDH).

Serum cytokine analysis
Blood samples from patients were analyzed in duplicates for quantitative measurement of serum cytokines using enzyme-linked immunosorbent assay (ELISA) kit: Bio-Plex Pro Human Cytokine 17-plex Assay (Biorad).
Statistical analysis
Descriptive statistics of continuous variables were expressed as medians with the interquartile range in parentheses. Continuous variables were compared between groups using the Mann-Whitney U test for nonparametric data. p value < 0.05 was considered to be statistically significant. All statistical analysis was performed using Graphpad Prism 5.03 for Windows, GraphPad Software, San Diego, California, USA.
Results
Clinical & laboratory characteristics
Patients were divided into groups based on diagnosis of NAFL or NASH by histological variables, or NAFLD patients with or without hepatic iron deposition (presence of stainable iron on liver biopsy samples). Across all patients, the mean patient age was 47 (± 9yrs) years, 49% were male, and 67% had a BMI greater than 30 kg/m2. There were more females (26/45) (p = 0.053) in the NASH group and NASH patients had higher AST (p = 0.01) and more severe histological scoring (steatosis, ballooning, inflammation and fibrosis, p < 0.0001) (Table 2). Elevated serum ferritin was present in 34% of the patients, 31% had diabetes, 52% had hypertension and 51% had hyperlipidemia.

Statistical significance (p < 0.05) was determined by Mann-Whitney U test.0
Table 2 Baseline characteristic, values are presented as the median (25th to 75th percentile) for physiologic variable, laboratory values and histological scores.
Analysis revealed that more individuals were males (19/26) (p = 0.008) in the cohort defined as NAFLD with hepatic iron. This group also had a significantly lower BMI (p = 0.03), CRP (p = 0.0093), total iron binding capacity (TIBC) (p = 0.0498), Alkaline phosphatase (p = 0.0128) and HDL (p = 0.0287), and displayed a higher HDL (p = 0.028), serum ferritin (p = 0.0009), serum iron (p = 0.0079), transferrin-iron saturation (p = 0.0005), serum albumin (p = 0.0052), AST (p = 0.001), ALT (p = 0.0045), total cholesterol (p = 0.039), triglycerides (p = 0.029), and LDL (p = 0.05) (Table 3).
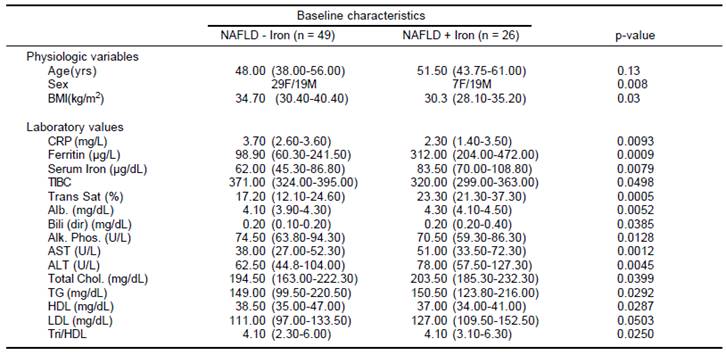
Statistical significance (p < 0.05) was determined by Wilcoxon Rank sum test, after adjusting for variables such as age, sex and BMI. NAFLD: nonalcoholic fatty liver disease.
Table 3 Baseline characteristic values of NAFLD patients with or without hepatic iron deposition are presented as medians (25th to 75th percentile) for physiologic variables and laboratory values.
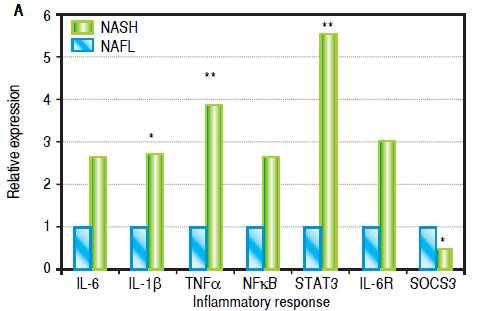
Increased hepatic gene expression of proinflammatory cytokines in patients with NASH relative to patients with NAFL
Levels of IL-1β (2.7 fold, p = 0.046), STAT3 (5.5 fold, p = 0.004) and TNFα (3.8 fold, p = 0.01) were upregulated in patients with NASH. While hepatic expression of IL-6, IL-6R and NFκB genes were also higher in NASH subjects, statistical significance was not achieved. SOCS3 expression was significantly reduced in patients with NASH (Figure 1A).
Hepcidin and TMPRSS6 expression is increased in patients with NASH
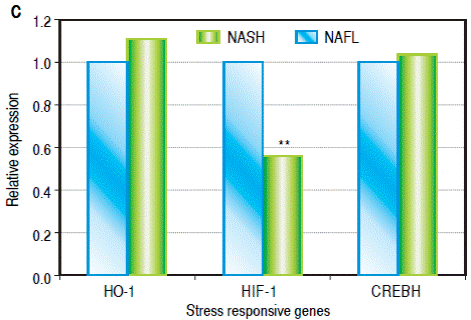
Hepatic gene expression levels of hepcidin (HAMP) were higher (2.3 fold, p = 0.02) in patients with NASH compared to NAFL subjects. Analysis of additional iron homeostasis genes within the BMP/SMAD pathway (HJV, FPN and BMP6) did not show significantly elevated expression profile in our patient population, even though HJV expression showed a trend towards increase (p = 0.06) (Figure 1B). Interestingly, we observed that the expression of TMPRSS6, a negative regulator of hepcidin expression, was higher in patients with NASH (8.4 fold, p = 0.003). We also observed a significantly decreased expression of HIF1α (p = 0.004), a redox sensitive gene, in patients with NASH. Expression of HO-1 and CREBH did not show any statistically significant alterations between the two groups (Figure 1C).

Figure 1B Hepatic mRNA expression of HAMP, HJV, FPN1, BMP6 and TMPRSS6in patients with NASH compared to patients with NAFLD. p values: HAMP: 0.02, HJV: 0.06, FPN: 0.22, BMP6: 0.28. TMPRSS6: 0.004.
Excess hepatic iron is associated with increased hepcidin and reduced inflammatory cytokine expression in NAFLD patients with stainable iron deposition
A significant increase in hepcidin expression (3.1 fold, p = 0.04) was observed in NAFLD patients with histological evidence of stainable hepatic iron compared to NAFLD patients without iron. Expression of IL-1β (4 fold, p = 0.06), TNFα (3 fold, p = 0.13), IL-6 (8 fold, p = 0.008), NFkB (5 fold, p = 0.02) and SOCS3 (2 fold, p = 0.07) were downregulated in NAFLD patients having hepatic iron overload relative to NAFLD patients lacking hepatic iron overload. Of these, hepatic gene expression of IL-6 and NF-κB were significantly reduced. Further, CREBH, a liver- specific transcription factor which is linked with ER (endoplasmic reticulum) stress, had lower expression (2.5-fold, p = 0.007) in NAFLD patients with iron overload. Expression of TMPRSS6 (p = 0.19) and STAT3 (p = 0.97) were higher in patients who had excess hepatic iron, but not statistically significantly so (Figure 2).

Figure 2 Effects of excess iron in patients with NAFLD. Hepatic mRNA expression of: A. Inflammatory markers IL-6, IL-1 β, TNFα, NFκB, STAT3, IL-6R and SOCS3 (p values; IL-6: 0.008, IL-1β: 0.06, TNFα: 0.13, NFκB: 0.02, STAT3: 0.97 IL-6R: 0.29, SOCS3: 0.07). B. Iron regulatory genes HAMP, HJV, FPN1 BMP6 and TMPRSS6 (p values; HAMP: 0.04, HJV: 0.16, FPN1: 0.51, BMP6: 0.47 and TMPRSS6: 0.19), and C. Stress-responsive genes CREBH, HO-1 and HIF-1α (p values; CREBH: 0.007, HO-1: 0.66 and HIF-1α: 0.35) in NAFLD + iron patients compared to NAFLD-iron patients. All values are shown as medians and statistical significance (p < 0.05) marked* and p < 0.01 is marked.**
HAMP expression strongly correlates with HJV expression
Linear regression analyses were performed between the various genes that were tested, and among them, the log mRNA expression of HAMP with HJV gene were observed to show a positive association in a statistically significant manner. As expected, the most significant correlation was found in NAFLD subjects with iron overload (r value = 0.718, p value < 0.0001) although a significant relationship was also found in all NAFLD subjects (r value = 0.652, p value < 0.0001) and all NASH subjects (r value = 0.669, p value < 0.0001) for HAMP with HJV (Figure 3).
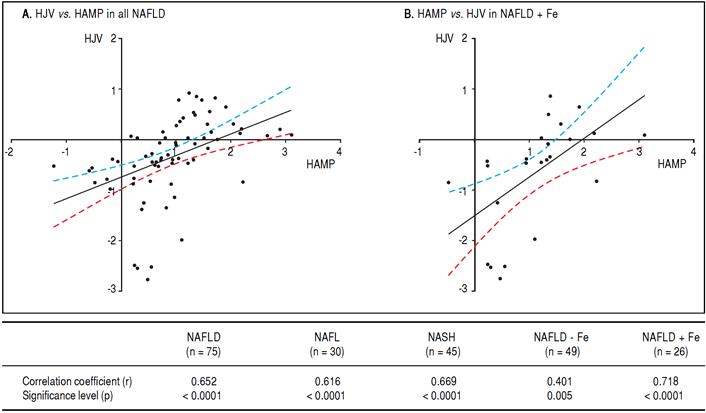
Figure 3 Linear regression in all NAFLD subjects (n = 75) are shown in (A) and in NAFLD patients with hepatic iron overload (NAFLD + Fe) (n = 26) (B). The regression line obtained with best fitted values and 95% confidence interval is shown. The table shows correlation coefficients between the log hepcidin mRNA and HJV mRNA in the various study groups. The r and p values are shown for each correlation and linear regression analysis. Since multiple analyses were performed, the cutoff was adjusted based on the Bon Ferroni equation to p < 0.01.
Serum cytokine analysis in patients with NASH relative to NAFL
Of all the cytokines and chemokines measured, IL-6 (0.7 vs. 3.5 ng/mL, p = 0.0001) and IL-8 (2.6 vs. 5.6 ng/ mL, p = 0.003) were significantly elevated in NASH patients relative to NAFL patients. Notably, there was a significant positive correlation between serum IL-6 cytokine and hepatic IL-6 gene expression (r = 0.572, p < 0.0067), and between serum IL-6 levels and hepatic hemojuvelin gene expression (r = 0.756, p < 0.0001) in NAFL patients, but not in NASH patients or NAFLD patients with or without iron overload. In addition, we observed a positive correlation between serum TNF-α levels and hepatic IL-6 gene expression levels (r = 0.602, p < 0.0038), and between serum TNF-α levels and hepatic HJV gene expression (r = 771, p < 0.0001) in NAFL patients, but not in NASH patients, or NALFD patients with iron or without overload. Finally, we observed a positive correlation between serum IL-8 levels and hepatic IL-6 gene expression levels (r = 0.051, p = 0.018), between serum IL-8 levels and hepatic HJV gene expression (r = 0.804, p < 0.0001) and in between serum IL-8 levels and hepatic CREBH gene expression (r = 0.497, p = 0.021) in NAFL patients, but not in NASH patients, or NAFLD patients with or without iron overload.
Discussion
The goal of this study was to identify differential expression of genes involved in iron regulation, inflammation and stress response in patients with NASH compared to NAFL. This information is crucial towards identifying factors that contribute to progression of fatty liver disease. Evidence continues to accumulate strengthening the clinicopathologic correlation between hepatic iron deposition and more specifically, its deposition pattern and progression of NAFL to NASH. Iron deposition within hepatic RES cells was found to be an independent predictor of worsening fibrosis and advanced liver disease (higher NAS score) in patients with NAFLD.5 Hepcidin is an important regulator of inflammation in the liver9,16 and along with its key role in iron homeostasis,6 it could be playing a vital part in NASH pathogenesis. We hypothesized that hepcidin and/or its upstream regulatory factors play a key role in progression of NAFL to NASH. Conflicting data exists regarding hepatic hepcidin expression in NAFLD where both a decreased20 and increased expression pattern has been reported10,21 compared to controls. In our patient population, hepatic hepcidin expression was significantly elevated in patients with NASH compared to NAFL. Taken together with the increased hepatic gene expression of inflammatory cytokines IL-6, IL-1 β and TNFα, it is plausible that there is increased hepcidin expression that is attributable to its stimulation by IL-6 through the JAK2/ STAT3 pathway.9,22-25 Furthermore, the reduced gene expression of HIF1α, a hypoxia-inducible negative regulator of hepcidin,9 could have also contributed to the upregulation of hepcidin expression in the NASH patient population.
Hepcidin expression was also found to be higher in patients with hepatic iron deposition which constitutes a physiological response of the liver to decreased circulating iron levels.15,16 High ferritin and low expression of hepatic inflammatory cytokines (IL-6; 8 fold, IL-1β; 4 fold, and NFκB; 5 fold) in patients with NAFLD containing hepatic iron deposition could possibly be suggestive of the notion that in this cohort the increased hepcidin expression is more likely attributable to hepatic iron deposition rather than inflammation. In our patient population, we did not observe upregulation of SOCS3, a known negative regulator of the JAK2/STAT3 signaling pathway26 and thereby of hepcidin, raising the possibility of other mechanisms distinct from SOCS3 being involved in suppression of inflammatory response. CREBH, a negative regulator of hepatic lipogenesis and a marker of ER stress, was reduced in NAFLD patients with hepatic iron deposition. This finding is consistent with previous reports that hepatic iron directly results in excess hepatic lipid load.
Similar to hepcidin, HJV is detected predominantly in hepatocytes.28,29 We found a strong association between hepcidin and hemojuvelin mRNA levels in NAFLD patients with hepatic iron deposition, reinforcing its role in hepcidin regulation. HJV plays a central role by interacting with BMP6, TMPRSS6 (also known as matriptase-2), and neogenin, in the regulation of hepatic hepcidin expression.30 Expression of TMPRSS6, a negative feedback inhibitor of hepcidin expression,11 was significantly higher in patients with NASH compared to NAFL. TMPRSS6 functions to suppress BMP-mediated stimulation of hepcidin transcription through cell surface proteolytic processing of the BMP co-receptor, HJV. Increased expression of TMPRSS6 in our NAFLD patient population likely represents a potential mechanism involved in the regulation of hepcidin.
Hepatic HAMP gene expression is induced in patients with NASH compared to NAFL, and presumably in response to hepatic iron excess in NAFLD patients with iron overload. Two possible mechanisms for hepcidin expression in patients with NASH are likely IL-6-mediated stimulation of JAK2/STAT3 pathway, which results in upregulation of hepcidin and reduced gene expression of HIF1α. Furthermore, increased hepatic STAT3 gene expression in NASH patients relative to NAFL patients lends support to this putative hypothesis.
Iron deposition in livers of patients with NAFLD can be hepatocellular, reticuloendothelial, or both. A study of 849 adult biopsy specimens, performed in the United States by us, observed that reticuloendothelial patterns of iron deposition was associated with advanced fibrosis compared with hepatocellular iron. Biopsy specimens with reticuloendothelial iron were also more likely to have definite steatohepatitis.5 However, an Italian study of 587 patients with NAFLD found that hepatocellular rather than reticuloendothelial iron was associated with an increased likelihood of fibrosis.31 The discordant results may be explained by differences in the patient populations; the subjects in the US study were more ethnically diverse and had higher body mass indices and more advanced fibrosis than those in the Italian study. Interactions between iron metabolism and NAFLD are complex and under active investigation by others and us.
In this study, we found increased expression levels of in hepcidin in both NAFLD patients with iron and NASH patients. In our studies, the increased hepcidin levels mitigate inflammatory responses through a different pathway other than the well-studied SOSC3 mechanism. Though there is an increase in hepcidin levels in NASH patients, which could be in response to hepatic iron or increase in IL-6/STAT3 or decrease in HIFa, or a combination of these influences, we believe that increase in TMPRSS6, a negative regulator of hepcidin, could have hindered the anti-inflammatory properties of hepcidin and contributed to the transition of NAFL to NASH. However, further studies are required to confirm the anti-inflammatory role of hepcidin, to identify the pathway by which it mitigates the inflammatory response and to identify the mechanism by which TMPRSS6 influences hepcidin expression and interferes with its anti-inflammatory effect in NASH patients. We propose that further studies in a larger independent cohort would increase our understanding of the pathogenesis of NASH and possible pharmacological solutions. Further, undertaking an unbiased approach and performing RNA-seq on NAFL/NASH/ NAFLD with iron overload populations will allow us to uncover novel regulatory proteins associated with this complex disease.
While this study has provided us with a number of important insights regarding the regulatory pathways such as iron homeostatic pathways, inflammation and stress responses that could be influencing NAFL to NASH pathogenesis and provided us with information about pathways that could be targeted, there are several limitations of the study, in that, we did not perform an a priori power calculation, since we had sufficient numbers of patients to perform a powered study. In addition, we did not record the intake of iron from both dietary sources and supplements. No information was collected on other medication and diseases that may affect iron metabolism. Our study is also limited by the lack of serum hepcidin measurements and HFE genotyping, which may have provided additional valuable insights into hepatic iron regulation in our patient population.
In conclusion, we observed that HAMP expression is elevated in NASH patients and in NAFLD patients with hepatic iron deposits. Our data allowed us to study the interdependence of various regulatory signals such as hepatic iron stores, inflammation and hypoxia or oxidative/ ER stress on the expression of hepcidin and inflammatory cytokines. It is likely that the increased hepcidin expression is a protective response, which attempts to sequester excess iron thereby reducing oxidative stress. This effect might also play a role in mitigating the inflammatory response as seen in our NAFLD patients with hepatic iron overload. Examination of hepcidin regulatory mechanisms in a larger patient population will help us to understand the pathogenesis of NASH and in turn help identify potential ‘druggable’ targets to halt its progression.

