











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
Chronic liver disease (CLD) and cirrhosis are major sources of morbidity and mortality in the United States as well as in Mexico.1-3 A recent publication1 point out the importance of CLD and cirrhosis among different ethnic groups in the United States. Non alcoholic fatty liver disease (NAFLD) was the most common cause of cirrhosis in the entire cohort. By ethnicity, NAFLD was the most common cause of cirrhosis in Japanese Americans, Native Hawaiians, and Latinos, accounting for 32% of cases. Also alcoholic liver disease was the major cause of cirrhosis in whites (38.2%), while hepatitis C virus was the most common cause in African Americans (29.8%).
Furthermore, it has been suggested that about 0.1% of the European population is affected by cirrhosis, corresponding to 14-26 new cases per 100,000 inhabitants per year or an estimated 170,000 deaths per year.4
Fibrosis in many organs is produced by a pathological excess in the deposit of extracellular matrix, including collagen types 1 and 2, along with chronic inflammation. In the liver, it is associated with the consumption of alcohol, NAFLD, viral or autoimmune hepatitis, and systemic diseases (Figure 1).5
The response of hepatocytes to inflammation plays a decisive role in the physiopathology of hepatic fibrosis, which involves the recruitment of both pro- and anti-inflammatory cells such as monocytes and macrophages. These amplify the response throughout the production of other cytokines and chemokines, which increase the stimulus of hepatic stellate cells by activating proinflammatory cells. Fibrogenic cytokines, such as transforming growth factor beta (TGF-β), activated by macrophages facilitate the transdifferentiation of stellate cells to myofibroblasts, which are the main source of production of extracellular matrix.6,7 Understanding these mechanisms has several clinical implications, including the development of therapeutic options that allow us to prevent, stop, or revert the progression of hepatic fibrosis and improve liver function (Figure 2).8 Unfortunately, there is no effective antifibrotic treatment approved for human use, and the point at which fibrosis becomes irreversible is not yet recognizable. The formation of regenerative nodules, the development of portal hypertension, and data indicating early hepatic failure are considered irreversible changes. However, it has been proved that in the early stages of fibrosis when the cause has been treated (e.g., hepatitis B or C), regression occurs in at least 70% of patients with the right antiviral management.9,10
The aim of this review is to identify the therapeutic options available for the treatment of this pathology, enabling the prevention of progression when it is detected in time.
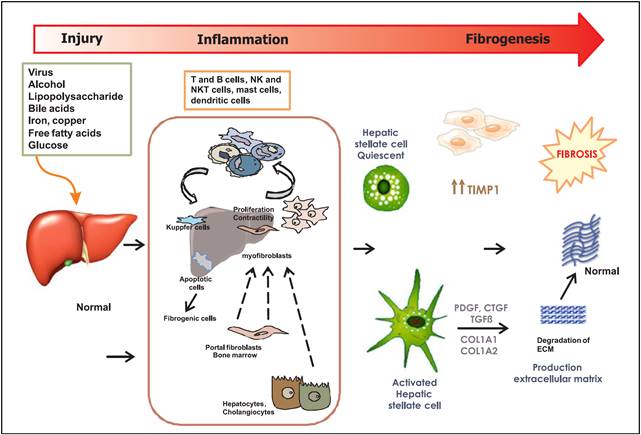
Figure 1 Mechanisms of hepatic fibrosis. Liver injury may be caused by multiples factors. These factors in turn induce liver inflammation through different types of cells. Also the oxidative stress, nitrosative stress, and apoptosis. Hepatic stellate cells are the key cells in the process of liver fibrosis. Some factors that regulate HSC activation, proliferation, function, and survival represent important therapeutic targets; likewise, therapies that directly degrade scar and/or promote liver regeneration. NK cells: NKT cells. PDGF: platelet-derived growth factor. CTGF: connective tissue growth factor. TGFβ, TGF-β: transforming growth factor-β. COL1A1: composed of collagen type I alpha 1. COL1A2: composed of collagen type I alpha 2
Therapeutic options
The therapeutic intervention points can be classed as: eliminating the stimulus or harmful cause; suppressing hepatic inflammation; interfering in the activation of stellate cells; and promoting the deterioration of extracellular matrix. The most effective way to prevent the formation of fibrosis is by eliminating the stimulus or harmful cause of hepatic damage, but this is not always feasible.
Steroids are most commonly used in treating many liver diseases. An example of this is in the histopathological improvement of liver swelling in patients with autoimmune hepatitis. In fact Czaja and Carpenter have reported a decrease in 53 % of fibrosis score of treated patients during 57 ± 7 months. Those researches also observed an improvement in their histological activity score (61 vs. 32%, P = 0.02). Additionally, they suggested that the conventional corticosteroid regimens are able to prevent, stabilize, or reverse hepatic fibrosis in up to 80% of patients.11 The molecular mechanisms through corticosteroids exert the anti-fibrotic actions have been reviewed recently by Montaño-Loza, et al.12
Briefly, the effects of corticosteroid therapy in autoimmune hepatitis are to limit tissue injury, reduce the molecular signals for fibrosis, and facilitate degradation of the extracellular matrix.
Regarding to the effect of caffeine on the liver fibrosis. It is known that caffeine is a purine alkaloid, acting through the antagonism of adenosine receptors A1 and A2A, with its main effect being observed at therapeutic concentrations of 10-100 μM. Interestingly Modi, 13 observed in 177 patients with liver fibrosis from different etiologies: 121/177 (68%) had chronic hepatitis C; the remaining patients had chronic hepatitis B (13%), delta hepatitis (3%), non-alcoholic steatohepatitis (11%), primary biliary cirrhosis (2%) or autoimmune hepatitis (3%) that daily caffeine consumption above the 75th percentile for the cohort (308 mg ~2.25 cups of coffee equivalents) was associated with reduced liver fibrosis (OR 0.33, 95% CI: 0.14-0.80, p = 0.015).
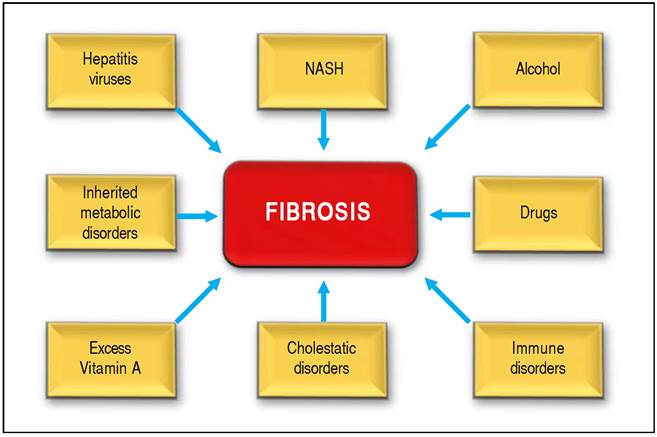
Figure 2 Shows several causes of hepatic fibrosis: viral infection is most common (hepatitis B and C). Nonalcoholic steatohepatitis (NASH). Also liver fibrosis is associated with the consumption of alcohol, or autoimmune hepatitis and chronic cholangiophaties.
In other study, Ruhl and Everhart14 found in the Third US National Health and Nutrition Examination Survey, 1988-1994 by using the ALT activity (> 43 U/L) as a marker of liver injury in subjects at high risk for liver injury (with excessive alcohol consumption, viral hepatitis, iron overload, overweight, or impaired glucose metabolism) elevated ALT activity was in 8.7% of this high-risk population. In unadjusted analysis, lower ALT activity was associated with increasing consumption of coffee (P = 0.001) and caffeine (P = 0.001). Multivariate logistic regression analyses showed that the risk of elevated ALT activity declined with increasing intake of coffee (P for trend = 0.034) and caffeine (P < 0.001). Comparing persons who drank more than 2 cups per day with non coffee drinkers, the odds ratio was 0.56 (95% confidence interval, 0.31-1.0). Comparing persons in the highest caffeine quintile with the lowest, the odds ratio was 0.31 (95% confidence interval, 0.16-0.61). We believed that the consumption of caffeine benefits patients with chronic liver disease possibly via the blockage of adenosine, a strong endogenous regulator of inflammation and tissue repair that limits the extent of fibrosis.15 Caffeine 2A receptors are expressed in stellate cells, and the activation of these receptors promotes collagen production. It is also important to mention that caffeine has an positive effect in patients with hepatic steatosis. In fact, we observed a dose-dependent reduction in the consumption of caffeine with increasing severity of steatosis in the general population. This effect has been observed in animal models of fatty liver, in which caffeine intake improves insulin resistance and reduces inflammatory cytokine production.16
Ursodeoxycholic acid (UDCA) is a physiologic hydrophilic dihydroxy bile acid.17 The antifibrotic properties of UDCA have been known for a long time. In fact UDCA is used for treatment of primary biliary cholangitis (PBC) in humans which improves clinical indices and helps delay liver transplantation.18 It is also observed that administration of UDCA to patients with PBC is associated with a considerable decrease in the progress of liver fibrosis. Corpechot, et al. assessed the effect of UDCA therapy on liver fibrosis progression in PBC by a Markov model the progression rates between early and late histologic stages in 103 patients with PBC enrolled in a randomized, double-blind, placebo-controlled trial of UDCA. UDCA therapy was associated with a 5-fold lower progression rate from early stage disease to extensive fibrosis or cirrhosis (7% per year under UDCA vs. 34% per year under placebo, P < 0.002), but was not associated with a significant difference in regression rates (3% per year under both UDCA and placebo). When those investigators assessed at 4 years, the probability of UDCA-treated patients to remain in early stage disease they found a 76% (95% confidence interval: 58%-88%), as compared with 29% (15%-52%) in placebo-group.19 The effect antifibrotic activity of UDCA is complex and involves replacement of endogenous cytotoxic bile acids [chenodeoxycholic (CDCA) and DCA acid] by UDCA a non-cytotoxic bile acid; the mechanism is likely to be competition for active ileal transport.17 Those changes in turn might induce inhibition of free oxygen radical-dependent processes in the liver and by reduction in production of cytokines, in particular of tumor necrosis factor-alpha (TNFα) and tumor growth factor-beta (TGFβ), inducing apoptosis and coordinating processes of inflammation and fibrogenesis.20,21 The UDCA has shown a beneficial effect, specifically in patients with PBC as we mentioned before.22 Furthermore, NorUDCA, a C23 homolog of UDCA that lacks a methylene group in its side chain is not accumulated in the enterohepatic circulation and does not cause hepatotoxicity after its chronic administration.23 NorUDCA has a clear advantage over its parent compound, UDCA, in improvement of liver pathology. In experiments with Mdr2-knockout mice having advanced cholestatic liver disease including sclerosing cholangitis and biliary fibrosis.24 The administration of norUDCA improved liver damage more effective than UDCA.25 In agreement with authors. We believed that norUDCA increased hydrophilicity that led to a decrease in its cytotoxicity and induction of detoxification pathways and elimination routes for bile acids.
Focusing on immunomodulators, natural killer (NK) cells are part of the innate immune system and represent 50% of the lymphoid hepatic reserve. Their primary function is in defense against infectious agents; however, they can diminish hepatic fibrosis through the inhibition or apoptosis of stellate cells via the retinoic acid pathway.26 The destruction of NK cells leads to the liberation of antifibrotic cytokines such as interferon-alpha (IFN-α) and IFN-gamma (IFN-γ), which suppress the transcription of genes involved in collagen production. IFN-α antagonizes the TGF-β/Smad3 pathway, which suppresses the activation of the collagen type I alpha 2 chain (COL1A2) gene, providing a molecular basis for antifibrotic effects.27 Jeong, demonstrated that the consumption of alcohol had a moderating effect on the antifibrotic effect of NK cells, which could help explain the accelerated formation of fibrotic tissue in patients with liver diseases caused by alcohol. 28 There are also studies using animal models where therapeutic options are proposed as ways to promote the survival of NK cells.29,30 However, these approaches have yet to demonstrate a benefit in human.
Other mechanisms thought to be involved in fibrosis involve ligands of receptors of farnesoid X (FXR), a nuclear transcription factor that is activated by conjugating to bile acids. Some studies show that CDCA is an endogenous ligand that plays an important role in the regulation of secretion and bile flow.31 Along with regulating hepatocyte activation, CDCA has shown an antifibrotic effect in animal models. The administration of 1-10 mg/kg of CDCA for 12 weeks led to a reduction in the expression of collagen type 1, TGF-β, as well as a lower production of tissue metalloproteinases in 90% of mice, together with an inhibition of fibrogenesis in 70-80% of them.32 Furthermore, Mudaliar,33 have reported that use of obeticholic acid (a semisynthetic derivative of CDCA) administered for 6 weeks at doses of 25-50 mg was well tolerated, sensitivity to insulin increased, and there was a decrease in inflammation in the liver and of fibrosis in patients with type 2 diabetes mellitus and fatty liver disease. It appears that this approach can improve the integrity of hepatocytes. Interestingly, the gut microbiota has recently been shown an effect the size and composition of the bile acid pool throughout the enterohepatic circulation via FXRdependent mechanisms.34 Furthermore, OCA can increase ileal FGF15 and suppress hepatic CYP7A1 expression in germ-free mice, these results suggest its capacity to reverse their reduced FXR signaling and a potential cross talk between OCA and the gut microbiota.
Endocannabinoids are lipid molecules that act on the cannabinoid receptors CB1 and CB2. An increase in the activity of these receptors is involved in the pathophysiology of visceral obesity and its complications,35 including diabetes mellitus type 2,36 and in the pathophysiology of fatty liver disease,37 and viral hepatitis.38 Activation of the CB1R receptor induces the expression of profibrotic genes and promotes the formation of hepatic fibrosis. Blocking such a receptor has an opposite effect. However, its inhibition has been linked to adverse neuropsychiatric side effects.
Inducible nitric oxide synthase also stimulates hepatic fibrosis.39 An increase in the activity of this enzyme contributes to the development of alcoholic liver disease,40 the progression of viral hepatitis,41 insulin resistance, and obesity. Cinar,7 introduced a peripheral antagonist of the oral receptor CB1R that accumulates in the liver, where it liberates an inhibitor of nitric oxide synthase. With this dual function, a decrease in the rate of progression of hepatic fibrosis and an attenuation of established fibrosis were found, suggesting a future therapeutic option. Another possible drug is Cenicriviroc, an oral antagonist of the chemokine receptors CCR2/CCR5, which showed an antifibrotic effect in animal models of hepatic and renal fibrosis.42 Currently, there is a phase 2b study (CENTAUR) being conducted on adult patients with NASH and hepatic fibrosis.
Accordingly, antioxidants also play a role in the management of fibrosis and most studies discuss the use of vitamin E and its effect on fibrogenesis. Despite the controversial effects. Sanyal,43 reported on a study in which 247 patients were randomized to receive pioglitazone (30 mg/day), vitamin E (800 U/day), or a placebo for 96 weeks. Those patients who received vitamin E presented statistically significant improvements in NASH (p = 0.001), and a reduction in aspartate transaminase (AST) levels. The study also found reductions in fatty liver disease (p = 0.005 for vitamin E and p < 0.001 for pioglitazone), improvements in lobular inflammation (p = 0.02 for vitamin E and p = 0.004 for pioglitazone); however, there was no significant amelioration in the fibrosis scales (p = 0.24 for vitamin E and p = 0.12 for pioglitazone).
Silymarin (from Silybum marianum) is a natural herbal component extracted from cardoon milk. This component has shown antifibrotic activity through the cytoprotection and inhibition of hepatic Küpffer cells. Trappoliere, et al.44 demonstrated that this agent had a dose-dependent inhibition on cell proliferation (p < 0.001), cell motility (p < 0.001), and the synthesis of extracellular matrix components (p < 0.05). In addition, it inhibited the synthesis of IL-1 and IL-8 (p < 0.01). A meta-analysis of 14 studies conducted by Jacobs, et al.45 found no significant effects on mortality reduction, nor any histological improvement.
Other compound that has been suggested as antifibrotic is the pirfenidone (PFD) (5 methyl-1-phenil-2 (1H)-pyridone) has proved antifibrotic and anti-inflammatory properties in a wide number of animal models of fibrosis. PFD effects are mediated in part through inhibition of NF-κ-B activation, these mechanisms included inhibition of PDGF, hepatic stellate cells (HSC) proliferation, reduction of TNF-α and IFN-α levels and decrease in iNOS/NO induction.46,47 Also, PFD down-regulates TGF-β1, TIMP-1, MMP-2 mRNA and collagen deposition.48,49
Flores-Contreras L, et al.50 Have reported that Pirfenidone for two years benefits chronic hepatitis C patients and improves inflammation, fibrosis and steatosis in higher number of patients as previously shown for 12-months treatment with PFD. Additionally, PFD improved TGFβ1 and IL-6 levels and diminished liver expression of anti-fibrogenic receptor CB2. Also it has been suggested that PFD has other potential effects to improve the liver fibrosis. However, we believe that more clinical studies are needed to provide solid evidence for a better understanding of mechanisms involved in the effects of anti-inflammatory and antifibrotic drugs.51
Other cells involved in the pathophysiology of fibrosis are dendritic cells (DC). Connolly, found that the depletion of these cells is related to an increase in the levels of inflammatory mediators responsible for the formation of Fibrosis in liver tissue.52 Dendritic cells represent 25% of hepatic leukocytes, so modulation of their function could be a therapeutic approach for hepatic fibrosis.53 Recently, Rahman and Aloman proposed a potential mechanism for the contribution of DC to fibrogenesis.
They stated that DCs regulate the number and activity of cells involved in the development of fibrosis (such as natural killer cells and CD8+ cells), and tissue remodeling, originally attributed to macrophages/monocytes, may be dependent on DC because of their ontogenetic relationship.54 On the other hand, it has been suggested that DC could play a role in the regression of liver fibrosis.55 Jiao, et al. induced liver injury in a murine model using carbon tetrachloride (CCl4) and evaluated fibrosis regression after cessation of insult in an environment depleted of DC.
Kluwe, et al. demonstrated a role for Jun kinase in inflammatory liver damage.56 Animal models have shown that to antagonize this enzyme might be an option for diminishing the formation of fibrosis. Other experimental anti-inflammatory options have been considered, such as the use of antagonists of lysophosphatidic acid (LPA), which plays a decisive role in multiple systemic diseases, including the formation of fibrosis.57
Furthermore molecules such as multimeric amyloid-P can modify the activation of macrophages attenuating inflammatory pathways. Castaño, et al.58 showed that this factor could stabilize inflammation through the local liberation of interleukin (IL)-10 in fibrotic kidneys of animal models. The antagonist of the protein galectin-3 is another therapeutic approach that is being studied, as this protein is critical for the development of hepatic fibrosis. Traber, et al. demonstrated that inhibiting its action for 8 weeks reduced the production of collagen by at least 50% in an animal model.59 In their study, hepatic biopsies revealed a decrease in the stage of fibrosis as well as a reduction in portal inflammation and ballooning.
Another group of treatments concerns hepatoprotective agents. These include hepatocyte growth factor (HGF), deletions or variations of this factor, and the use of synthetic HGF, growth factors similar to insulin, and caspase inhibitors. HGF shows antifibrotic activity by suppressing the action of TGF-β, inducing the expression of collagenases and inhibiting the apoptosis of stellate cells.60 Other studies in animal models support this antifibrotic effect.61 Caspase inhibitors represent another group of potential therapeutic agents. Hepatocyte apoptosis is an inflammatory profibrogenic process that increases in patients with alcoholic hepatitis and nonalcoholic fatty liver disease, and it correlates with the severity of the disease and fibrosis. Molecules that block caspases involved in the apoptotic pathway have been developed.62 However, because it has been reported that there might be an increase in the risk of malignancy, some studies with these pharmaceuticals have been stopped. No studies have demonstrated human toxicity. Canbay, et al. found that application of the caspase inhibitor IDN-6556 diminished the incidence of hepatic fibrosis in mice, with lower levels of alanine transaminase (ALT) and liver inflammation.63
Several studies have examined the use of angiotensinconverting enzyme inhibitors. Found in stellate cells, these receptors increase cell proliferation and the release of reactive oxygen species when activated, which stimulates the secretion of fibrogenic cytokines.64 Experimental studies in rats conducted by Yoshiji65 demonstrated that the antagonists of such receptors showed antifibrotic activity. Generally, they are well tolerated, representing a good therapeutic option.
TGF-β is the main cytokine implied in fibrogenesis. When its receptors are blocked, there is an inhibition in the production of extracellular matrix and an acceleration in its degradation. Studies on animals have already proven its benefits. Friedman66 demonstrated the fundamental role of this cytokine in hepatic diseases as well as the possibility of reverting fibrosis in early stages. Studies on curcumin have shown that it suppresses the expression of genes encoding TGF-β receptors in activated stellate cells via activation of peroxisome proliferator-activated receptor (PPAR)-gamma, which leads to an interruption in the signaling of TGF-γ.67 Because of concern that inhibiting this tumor suppressor pathway might increase the risk of developing neoplasms, other treatments have been developed with good results, such as the use of integrin antagonists. Wang, 68 that the alpha V beta 6 integrin is linked to fibrogenesis by the activation of TGF-β. Blocking this integrin in mice and humans diminished the progression of fibrosis. Studies on other integrins such as alpha V have shown that pharmacological blocking of this subunit mitigated the development of hepatic and renal fibrosis.69
Antagonists of endothelin receptors are implicated in possible therapies The most studied example is Bosentan, which diminishes the activation of stellate cells and the levels of collagen type 1 protein and of fibronectin mRNA from liver tissues.70 However, besides showing antifibrotic effects in experiments, the risk of hepatotoxicity discourages its regular use.
Cytokine antagonists belong to another pharmacological group formed by the blockers of platelet-derived growth factor (PDGF), blockers of the receptors of vascular endothelial growth factor (VEGF), intracellular cAMP modulators or in some transporters. Yoshiji,71 used an animal model employing double blockage of PDGF and TGF-β; applying Imatinib and angiotensin inhibitors showed a reduction in the development of hepatic fibrosis. Another example is Sorafenib, a multikinase inhibitor used on patients with hepatocellular carcinomas, which has shown antifibrotic effects in studies with animal models. Hennenberg, 72 showed that rats with hepatic damage administered 60 mg/kg per day for 1 week demonstrated a reduction in the formation of procollagen type 1 alpha, less deposit of perisinusoidal extracellular matrix, and minor activation of stellate cells, leading to lower intrahepatic resistance and reduced hepatic damage. Imatinib is commonly used in the management of leukemia and mesenchymal tumors, and has been shown to diminish hepatic fibrosis. It acts by antagonizing the tyrosine kinase receptor. Yoshiji, et al.73 used an animal model and found a dosedependent effect of Imatinib on reducing the proliferation, migration, and activation of stellate cells. However, another study only found improvements on an early stage of fibrosis with a reduction in the formation of extracellular matrix by 30% (p = 0.0455).74 Because the treatment was not effective on established fibrosis, Imatinib could only be a therapeutic option for early stages of fibrosis.
Matrix degradation is another useful therapeutic objective for organs with a certain stage of fibrosis. Using an animal model, Parsons, 75 demonstrated that a reduction in the degradation of extracellular matrix arose secondary to the expression of tissue inhibitor of metalloproteinase inhibitors (TIMP)-1. TIMP-1 promotes the survival of stellate cells that are key to matrix formation. When blocking the activity of TIMP-1, there was histological evidence of a reduction in the accumulation of collagen. Another study has focused on increasing the degradation of matrix histologically; however, its effectiveness has not yet been proven in human studies.76
Further studies are centered on trying to decrease the activity of stellate cells and to place them in a resting state. Two important studies by Troeger, and Kisseleva, showed in animal models that it is possible to revert the effect of fibrosis by inhibiting the function of stellate cells; however, the effects were still dependent on the implemented agents because the stellate cells reactivated when treatments were stopped. Consequently, ways of stopping the activation of these cells for the long term are still being studied.77,78
There are proposals of models in which apoptosis of stellate cells is encouraged for preventing fibrosis; however, it has not been possible to perform studies in which the loss of cells does not affect functional capacity.79 One study on gliotoxin, a fungal metabolite, proved to stimulate the apoptosis of stellate cells in vitro in an animal model. 80 This showed a histological reduction in the stage of the fibrosis thanks to the decreasing activity of stellate cells; nevertheless, there was no improvement on the hepatic function observed. Trebicka, et al.81 conducted an important study with atorvastatin, a statin, in which a dose of 15 mg/kg a day proved to reduce early fibrosis as well as the activation of stellate cells in an animal model. Although a long-term effect was not substantiated, there was a reduction in the expression of profibrotic cytokines, which enabled stellate cells to remain in a resting state with less proliferation and apoptosis. However, there is no evidence from human models to suggest the possibility of clinical recovery with this drug.
Conclusions
A number of therapeutic approaches—many of them still being researched—promise a way to prevent, stop, or revert the progression of hepatic fibrosis. The mechanisms must be clearly understood so that we can establish multidisciplinary approaches (combining mechanisms) to achieve long-term effects safely and efficiently. In the future, models of such combined strategies must be created to establish their safe use in humans. Although there are many therapeutic options, the optimal treatments currently remain unknown. We must also bear in mind that the management of affected patients might need to be individualized, taking into account specific types of liver fibrosis.

