











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
La estructura genética de una población, trata tanto de las frecuencias alélicas de los marcadores moleculares (o frecuencias nucleotídicas de las secuencias de ADN) y el número de grupos genéticamente divergentes en una población como variables aleatorias. Sin embargo, los análisis y comparaciones de los modelos pueden realizarse también usando un número fijo de grupos genéticamente divergentes o estructuras de poblaciones previamente especificadas (Corander et al., 2013).
Actualmente se ha utilizado el análisis Bayesiano de la Estructura Poblacional con el programa BAPS, por sus siglas en inglés, el cual puede agrupar datos moleculares y realizar análisis de mezcla genética y adición. El análisis de mezcla genética puede hacerse en (1) grupos (por lo general corresponde al agrupamiento de las poblaciones de la muestra), o (2) individuales. De hecho, en muchas aplicaciones con datos moleculares relativamente escasos conviene hacer análisis Bayesianos, cuando existe información auxiliar biológicamente relevante para definir los grupos antes del análisis de la mezcla. Ambos tipos de análisis de la mezcla genética pueden hacerse mediante (1) modelos no-espaciales, o (2) modelos espaciales, de discontinuidades genéticas en las poblaciones (Corander et al., 2013).
Hranitz y Baird (2000), encontraron que pocas poblaciones entre manchones de hábitat en el sitio de estudio de Crotaphytus collaris (Say) fueron suficientes para inferir divergencias genéticas o subdivisiones dentro de la metapoblación de estudio. El tamaño poblacional efectivo observado fue reducido y apoyó los modelos de evolución para C. collaris, donde los procesos estocásticos pudieron haber influido fuertemente en la divergencia de las poblaciones de esta especie.
Castillo-Ramírez et al. (2012), quienes estudiaron la vinculación de eventos fundadores con variación regional en recombinación en bacterias usando BAPS, en una gran colección mundial de secuencias de ADN de un genoma completo y datos derivados de las muestras, identificaron varios linajes genéticamente aislados dentro del Clon ST239 y estimaron sus tiempos de introducción en regiones geográficas particulares. Además, se demostró que dentro de un mismo país, el aislamiento geográfico de una zona a otra, tiene consecuencias en la medida en que la recombinación afecta la evolución genómica (Cheng et al., 2013).
Mulvaney et al. (2005), evaluaron la estructura de las poblaciones dentro de la especie del lacertilio Rhineura floridana (Baird) mediante la combinación de la distribución actualizada de datos con patrones geográficos utilizando genes del ADN mitocondrial (ADNmt). Encontraron una estructura genética poblacional bien definida combinando datos ADNmt y datos morfométricos.
Uma exsul (Schmidt y Bogert) es una especie microendémica restringida a depósitos de arena en el desierto Chihuahuense (≈500 km2), por esta razón se incluye en la lista roja UICN (Vázquez-Díaz et al., 2007) y en la NOM-059-SEMARNAT-2010 como en peligro de extinción (SEMAR-NAT, 2010). Es importante determinar la estructura genética poblacional de U. exsul para localizar y cuantificar dichas variedades genéticas mediante el análisis Bayesiano. Además, es necesario determinar las restricciones en el flujo genético y sus causas para fundamentar la importancia de cada una de las poblaciones, suponiendo que los grupos con distribución alopátrica, constituyen linajes distintos. Actualmente el cambio de uso de suelo y las actividades antrópicas siguen alterando y fragmentando el hábitat donde se distribuyen las poblaciones de U. exsul, por lo cual es importante conservar el germoplasma de todas sus variedades genéticas In situ.
Materiales Y Métodos
Trabajo de campo
El estudio se realizó tratando de abarcar todas las regiones de dunas del suroeste del Estado de Coahuila, en donde se ha reportado la distribución de U. exsul (Schmidt y Bogert, 1947; Gadsden et al., 2001; García de la Peña et al., 2005). Se delimitaron las poblaciones de la especie y se realizaron 14 salidas al campo durante los años 2016 y 2017, donde se muestrearon cinco individuos en cuatro poblaciones (Bilbao, Saucillo, Gabino, Texas) y cuatro individuos en la población de Estación Marte, para un total de 24 muestras (Figura 1). La captura se realizó manualmente mediante la técnica del lazado corredizo, se registró la fecha y las coordenadas geográficas de cada punto de colecta.
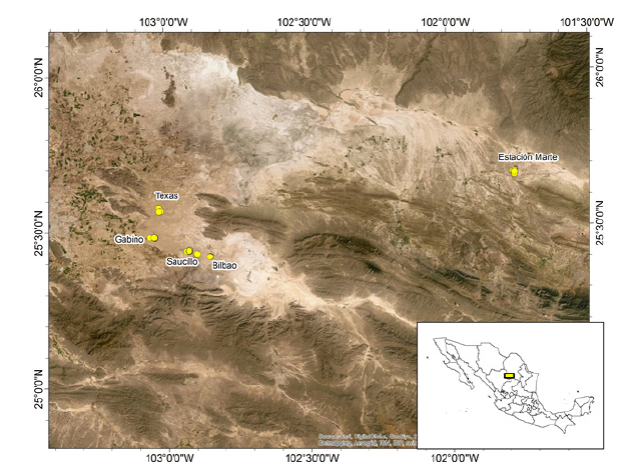
Figura 1 Área de estudio y puntos de colecta de las poblaciones de
Uma exsul.
Figure 1. Study
and sampling areas of Uma exsul population.
La colecta de material biológico se obtuvo In situ, un fragmento (~1cm) de tejido caudal de cada individuo capturado (sin lastimar al organismo) y se almacenó en un tubo de 1.5 ml con el Buffer de colección del Kit XpeditionTM Tissue and insect DNA MiniPrep de ZYMO RESERCH. CA, USA. Las muestras se transportaron al Laboratorio de Biología de la Conservación de la Facultad de Ciencias Biológicas (UJED), en donde fueron conservadas a -20°C hasta la extracción de ADN. Una vez tomada la muestra caudal de los individuos, estos fueron liberados vivos en ese momento en el punto exacto en donde fueron capturados. Tanto la pinza de corte como la región caudal del individuo fueron desinfectadas con alcohol al 90° GL.
Análisis de ADN genómico
Extracción de ADN. La extracción de ADN genómico se realizó a partir de tejido caudal de U. exsul mediante el Kit XpeditionTM Tissue and insect DNA MiniPrep de ZYMO RESERCH. CA, USA, siguiendo las instrucciones del proveedor. La concentración y pureza del ADN de las muestras fueron obtenidas en un espectrofotómetro (UV Transilluminator UVP®) y los productos de extracción de ADN fueron visualizados por electroforesis en geles de agarosa (Miller et al., 1988; Sambrook, 2001).
Amplificación de ADN. La amplificación del mtADN se realizó con los oligonucleótidos 5´ CCA TCC ACC ATC TCA GCA TGA TGA AA 3´ y 5´GCC CCT CAG AAT GAT ATT TGT CCT CA 3´ (Kocher et al., 1989; Fu, 2000; Murphy et al., 2006). Se utilizó el protocolo de PCR empleando 12.5 µL de MASTER MIX (PROMEGA®), Madison, USA, 1 µL de cada oligonucleótido (10 uM), 5 µL de ADN (50 ng totales) y 5.5 µL de H2O grado molecular. El programa térmico para el Cytb fue un ciclo inicial a 98 °C por 30 segundos, seguido por 33 ciclos de 5 segundos a 98 °C, 10 segundos a 65 °C y 30 segundos a 72 °C, y un ciclo final a 72 °C durante 5 minutos (Murphy et al., 2006). La amplificación se llevó a cabo en un termociclador (Thermo Scientific Piko 24®, CA, USA).
Visualización de los Productos de PCR. Los productos de extracción, así como los obtenidos de la amplificación y de restricción, fueron separados en geles de agarosa al 0.8 y 1.5%, respectivamente. Se utilizó como amortiguador TBE al 0.5% (tris base, ácido bórico, EDTA al 0.5M, pH 8.0). Las muestras fueron teñidas con Gel Red®, de acuerdo con el manual de Sambrook (1989) y se empleó un marcador de peso molecular de 100 pb (CSL-MDNA Cleaver Scientific Ltd. Camberley, UK) como referencia. Posteriormente cada uno de los geles fueron fotografiados con el fotodocumentador GelMaxTM UVP®. Los productos de PCR se enviaron a MACRO-GEN USA CORP. Rockville, Maryland, USA, para su purificación y posterior secuenciación.
Obtención de los datos genéticos
Las secuencias de nucleótidos fueron leídas y editadas con Chromatogram Explorer v. 5.0.2.3 (Heracle Biosoft SRL, 2006). Las secuencias se alinearon con ClustalW en Mega7 (Kumar et al., 2016). Se llevó a cabo el análisis de polimorfismo del ADN en el programa DnaSP v.5.10.01 DNA sequence polymorphism, de sus siglas en inglés (Librado y Rosas, 2009) para los siguientes parámetros: diversidad nucleotídica (π), la diversidad haplotípica (Hd), la D de Tajima para determinar la selección o deriva de las poblaciones y para determinar el estado poblacional en cuanto a expansión o estabilidad. Además, la lista y dispersión de los haplotipos para establecer la identidad de cada haplotipo en la red. Se implementó una AMOVA mediante el programa PopArt 1.7 (Population Analysis with Reticulate Trees) de sus siglas en inglés (Clement, 2002), para determinar la variación genética poblacional (Yoke et al., 2006). Se usó el programa DNAsp 5.10.01 para obtener diversas medidas de la heterogeneidad genética entre las poblaciones: Kst, Kxy y Fst (Hudson et al., 1992), con pruebas de permutación (aleatorización) de 1000 réplicas según Hudson et al. (1992a). También se utilizó el programa DNAsp 5.10.01 para realizar un análisis de dispersión “mismatch”. El cual permite la discriminación entre las poblaciones que se mantuvieron estables en el tiempo (curvas multimodales) y la población que experimentó una expansión demográfica desde una pequeña población fundadora (curvas unimodales).
Se utilizaron los programas TCS (Clement et al., 2000) y tcsBU (Santos et al., 2015) para establecer el arreglo de las relaciones haplotípicas entre poblaciones. El tcsBU es un programa informático online que se utiliza para estimar las genealogías genéticas, incluidas las multifurcaciones y/o las reticulaciones (es decir, las redes). Primero se estimó la red de haplotipos en el programa TCS v. 1.21 (Phylogenetic network estimation using statistical parsimony) de sus siglas en inglés (Clement et al., 2000) y este archivo se utilizó en el tcsBU para estimar la red de haplotipos. Las redes de parsimonia estadística fueron construidas o implementadas en TCS (Templeton et al ., 1992). Se utilizó el programa Arlequin (Scoffier, 2015) para determinar el haplotipo ancestral. Se utilizaron las secuencias del gen Citocromo b para realizar el análisis Bayesiano de la estructura genética poblacional, utilizando el software BAPS, el cual determina cuántos grupos genéticos hay en la población y cuántos grupos forman la población ancestral (Corander et al., 2013).
Resultados Y Discusión
Estadísticos descriptivos
Se obtuvieron fragmentos de ADN de 305 pb de las secuencias parciales del gene Citocromo b de U. exsul. El análisis de las secuencias mostró 48 sitios polimórficos y 22 haplotipos con diversidad haplotípica alta (Hd = 0.98). La diversidad nucleotídica de las cinco poblaciones juntas fue muy baja (n = 0.00187), la D de Tajima (D = 0, p ≤ 0.001) indica una baja selección y una población en expansión según el equilibrio de mutación-deriva (Tajima, 1989) y el análisis de distribución Mismatch (Figura 2). Lo anterior indica que en las poblaciones estudiadas opera la deriva génica. Sin embargo, analizando la población más divergente, que es la de Estación Marte, con un número promedio de divergencia nucleotídica de K = 23 (según el estadístico K de Jukes y Cantor, 1969) comparada con las demás muestras (Bilbao K = 5.5, Saucillo K = 8.1, Gabino K = 4.8 y Texas K = 4.1), se observa una diferencia relativamente alta que podríamos suponer que Estación Marte es una variedad genética diferente. En esta población (Estación Marte) se encontró una D de Tajima negativa (-2.35911, p < 0.001), esto puede indicar que esta población en particular tuvo un barrido selectivo reciente y posterior expansión de la población después de un cuello de botella reciente. El AMOVA aplicado a todas las poblaciones, indica que la variación interpoblacional es de 12.91 % la cual se considera baja, en cambio la variación intrapoblacional es alta (87.09 %), con Fst: 0.13113 y Nm: 3.31 (p < 0.001). Lo anterior significa que existe incipiente estructura genética y alto flujo genético interpoblacional. Sin embargo, si se considera la diferenciación genética entre poblaciones, a través de las distancias genéticas, se puede observar que la población de Estación Marte está muy alejada de las demás, con diferencias en distancias genéticas muy altas (Tabla 1). Por lo que se puede suponer que el alto flujo génico es de las poblaciones del Oeste (Bilbao, Saucillo, Gabino y Texas). Estación Marte está muy alejada geográficamente (155 km) de las demás poblaciones; por lo que podría considerarse una población alopátrica, es decir, una población aislada geográficamente de las demás poblaciones (Cabej, 2012). Además de que existe la barrera de la distancia, también las dunas están fragmentadas entre las poblaciones de oeste y la población de Estación Marte, por lo que se estima que no hay flujo génico entre estas poblaciones.
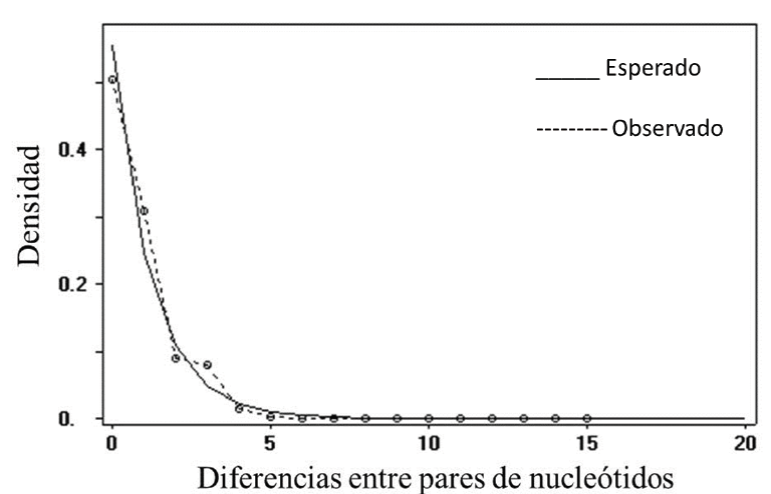
Figura 2 Distribución Mismatch para haplotipos del Citocromo B en Uma exsul de
las dunas de Viesca, Coahuila, México. La frecuencia esperada se
basa en un modelo de crecimiento-disminución de la población,
indicando aquí una expansión poblacional.
Figure
2. Cytochrome B haplotypes mismatch distribution in Uma exsul
of Viesca dunes, Coahuila, Mexico. Based on a model of population
growthdiminution, the expected frequency indicates a population
expansion.
Tabla 1 Diferenciación genética entre poblaciones a través de las distancias genéticas, donde:
Ks = número de sustituciones sinónimas por sitio, Kxy = número
promedio de sustituciones nucleotídicas por sitio entre poblaciones,
Fst = índice de fijación Haplotípica.
Table
1. Genetic differentiation between populations through
genetic distances, where: Ks = number of synonymous substitutions
per site, Kxy = average number of nucleotide substitutions per site
between populations, Fst = rate of fixing haplotype.
| Población 1 | Población 2 | Ks | Kxy | Fst |
|---|---|---|---|---|
| Bilbao | Gabino | 5.15 | 5.96 | 0.13591 |
| Bilbao | Marte | 13.27 | 14.9 | 0.04362 |
| Bilbao | Saucillo | 6.8 | 5.92 | -0.14865 |
| Bilbao | Texas | 4.8 | 8.56 | 0.43925 |
| Gabino | Martes | 12.88 | 15.9 | 0.12579 |
| Gabino | Saucillo | 6.45 | 7.8 | 0.17308 |
| Gabino | Texas | 4.45 | 5.96 | 0.25336 |
| Marte | Saucillo | 14.72 | 15.65 | 0.00639 |
| Marte | Texas | 12.5 | 16.4 | 0.17378 |
| Saucillo | Texas | 6.1 | 9.8 | 0.37755 |
Patrón de haplotipos
Se registraron 22 haplotipos de 24 secuencias del gen Citocromo b del mtADN de U. exsul, el haplotipo cinco (B5), agrupó 3 secuencias: B5, G1 y S3 (Tabla 2). Al respecto, Trepanier (2002) registró 12 haplotipos para esta especie, es de suponerse que las diferencias en el número de haplotipos encontrados, se debe a la selección de los sitios de muestreo donde incluyó un número mayor de muestras en sitos muy cercanos entre sí (población de Saucillo). Además, no incluyó la población de Estación Marte y por último, en este trabajo se utilizaron diferentes primers para amplificar el gen Citocromo b, ya que Trepanier (2002) llevó a cabo una metodología basada en Haddrath (Murphy et al., 2006), donde utilizaron un primer que no es específico para el Citocromo b mitocondrial, el cual ha sido utilizado comúnmente en aves. Lo anterior se comprobó con el programa BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Tabla 2 Distribución de haplotipos considerando una alineación de sitios con gaps, el cual
presentó 305 posiciones con 48 sitios polimórficos, 22 haplotipos y
una diversidad Haplotípica de 0.98. Donde, Hap_5: 3 [5-6 17] está
representado por tres haplotipos, 5, 6 y 17, que representan a los
haplotipos Bilbao5 (B5), Gabino1 (G1) y Saucillo3 (S3).
Table 2. Haplotype distribution,
considering an alignment of sites with gaps, which presented 305
positions with 48 polymorphic sites, 22 haplotypes and a haplotype
diversity of 0.98. Hap_5: 3 [5-6 17] is represented by three
haplotypes, 5, 6 and 17, representing the haplotypes Bilbao5 (B5),
Gabino1 (G1) and Saucillo3 (S3).
| Número de haplotipos | Nombre de haplotipos |
|---|---|
| Hap_1:1 [1] | Hap_1:1 [B1] |
| Hap_2:1 [2] | Hap_2:1 [B2] |
| Hap_3:1 [3] | Hap_3:1 [B3] |
| Hap_4:1 [4] | Hap_4:1 [B4] |
| Hap_5:3 [5-6 17] | Hap_5:3 [B5 G1 S3] |
| Hap_6:1 [7] | Hap_6:1 [G2]c |
| Hap_7:1 [8] | Hap_7:1 [G3] |
| Hap_8:1 [9] | Hap_8:1 [G4] |
| Hap_9:1 [10] | Hap_9:1 [G5] |
| Hap_10:1 [11] | Hap_10:1 [M1] |
| Hap_11:1 [12] | Hap_11:1 [M3]c |
| Hap_12:1 [13] | Hap_12:1 [M4] |
| Hap_13:1 [14] | Hap_13:1 [M5] |
| Hap_14:1 [15] | Hap_14:1 [S1] |
| Hap_15:1 [16] | Hap_15:1 [S2] |
| Hap_16:1 [18] | Hap_16:1 [S4] |
| Hap_17:1 [19] | Hap_17:1 [S5] |
| Hap_18:1 [20] | Hap_18:1 [T1] |
| Hap_19:1 [21] | Hap_19:1 [T2] |
| Hap_20:1 [22] | Hap_20:1 [T3] |
| Hap_21:1 [23] | Hap_21:1 [T4] |
| Hap_22:1 [24] | Hap_22:1 [T5] |
El análisis del patrón de haplotipos muestra tres grandes grupos y se reconoce a los haplotipos de Bilbao5, Gabino1 y Saucillo3, como haplotipos ancestrales (Figura 3). Sin embargo, en análisis con el programa Arlequín (Scoffier, 2015) se registró a Bilbao5 como el haplotipo ancestral (N = 24, p < 0.05), entonces se puede inferir que entre los tres haplotipos conforman el grupo haplotípico ancestral. El haplotipo M5 está muy alejado del resto con cuatro mutaciones, M1 y M3 son dos haplotipos desconectados del resto, lo que puede estar indicando que pertenecen a una posible variedad genética diferente. M4 está dentro de la red y conectado a los haplotipos que conforman la población de Texas. Lo anterior coincide con lo encontrado por Murphy et al. (2006) en donde encontraron que Uma scoparia Cope presentaba haplotipos únicos en parches de dunas muy alejados a los demás, lo cual daba la posibilidad de adaptaciones regionales y una posible especiación incipiente. Se observaron relativamente altas divergencias genéticas en las poblaciones de Estación Marte, mientras que la estructura genética dentro de las poblaciones del Oeste (Bilbao, Saucillo, Gabino y Texas) es menos discreta y con una divergencia muy ligera. Tomando en cuenta la distancia geográfica que existe entre la población de Estación Marte y el resto de las poblaciones y que los haplotipos de Estación Marte son los más alejados con dos haplotipos desconectados del resto, podríamos suponer que dicha población podría considerarse como una unidad evolutiva distinta (Figuras 3 y 4). Se recomienda realizar análisis morfológicos y de variabilidad genética intraespecífica de las distintas poblaciones adicionalmente a los resultados presentados en este trabajo.
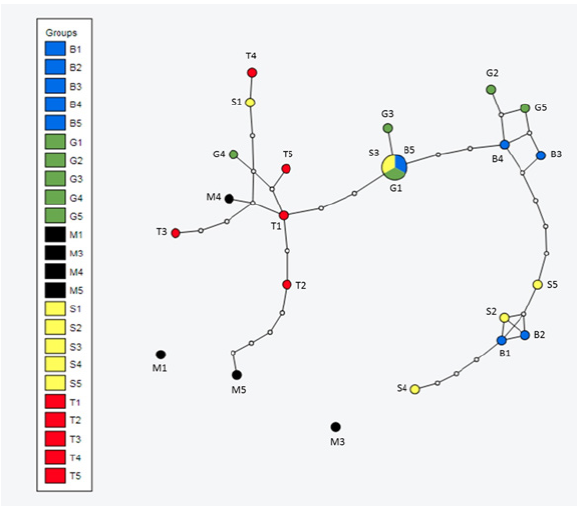
Figura 3 Patrón de haplotipos del gen citocromo b de las poblaciones muestreadas de la especie
Uma exsul, donde: B1 hasta B5 corresponden a
las secuencias de Bilbao, S1-S5 Saucillo, G1-G5 Gabino, T1-T5 Texas
y M1-M5 Marte.
Figure 3. Cytochrome b
gene haplotypes pattern of the Uma exsul species sampled
populations, where: B1 to B5 correspond to sequences of Bilbao,
S1-S5 Saucillo, G1-G5 Gabino, T1-T5 Texas and Mars M1-M5.
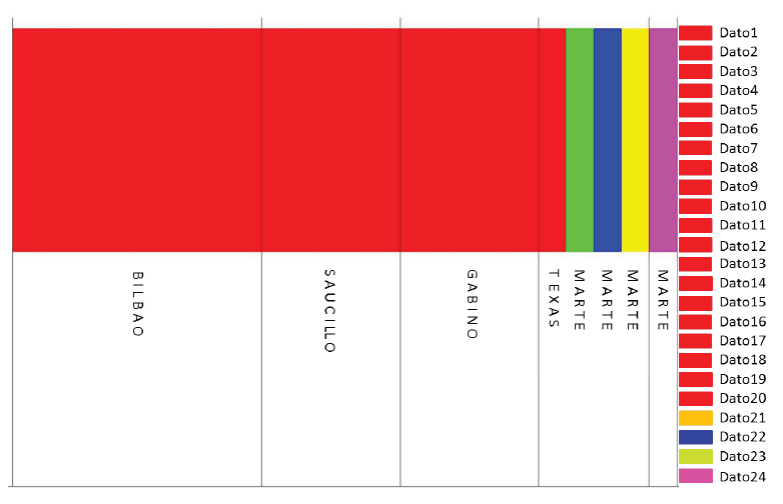
Figura 4 Estructura genética poblacional de Uma exsul mediante BAPS, donde: B1
hasta B5 = dato1-dato5 corresponden a las secuencias de Bilbao,
S1-S5 = dato6-dato10 corresponden a Saucillo, G1-G5 = dato11-dato15
corresponden a Gabino, T1-T5 = dato16-dato20 corresponde a Texas y
M1-M5=dato21-dato24 corresponden a Marte. Cada color es un grupo
genética-mente diferenciado y cada bloque (entre barras negras) es
un aislado. M5 (dato24) es un grupo diferente y aislado.
Figure 4. Population genetic structure of Uma exsul
using BAPS, where: B1 to B5 = data1-data5 correspond to Bilbao
sequences, S1-S5 = data6-data10 correspond to saucillo, G1-G5 =
data11-data15 correspond to Gabino, T1-T5 = data16-data20
corresponds to Texas and M1-M5 = data21-data24 correspond to Mars.
Each color is a genetically distinct group and each block (between
black bars) is an isolate. M5 (data24) is a different and isolated
group.
Figure 4. Population genetic
structure of Uma exsul using BAPS, where: B1 to B5 = data1-data5
correspond to Bilbao sequences, S1-S5 = data6-data10 correspond to
saucillo, G1-G5 = data11-data15 correspond to Gabino, T1-T5 =
data16-data20 corresponds to Texas and M1-M5 = data21-data24
correspond to Mars. Each color is a genetically distinct group and
each block (between black bars) is an isolate. M5 (data24) is a
different and isolated group.
Estructura genética poblacional
El análisis de estructura genética poblacional muestra cinco colores, cada color representa uno de los grupos genéticamente diferenciados y cada barra de color vertical corresponde a un aislado (Figura 4). El código de color es el siguiente: grupo BAPS1, Rojo (Bilbao, Saucillo, Gabino y Texas) el cual se denominó “Bilbao”; grupo BAPS2, Rojo, Verde, Azul y Amarillo (Texas) el cual se denominó “Texas”; grupo BAPS3, Rosa (Marte) el cual se denominó “Estación Marte”. Se encontraron pruebas de mezcla entre los dos primeros grupos y las muestras de M1, M3 y M4, representan grupos diferentes, pero enlazados con T5 (población de Texas). M5 de Estación Marte representa un grupo genético diferente y aislado, totalmente separado de los demás grupos de la estructura genética, que podría representar una posible nueva unidad evolutivamente significativa. Mulvaney et al. (2005) mencionan que la lagartija gusano Rhineura floridana Baird, presenta una estructura poblacional genéticamente diferenciada en grupos y encontraron que las poblaciones más aisladas geográficamente constituyeron grupos que representan especies diferentes. Por otro lado, Branch et al. (2003) y Heath et al. (2012) también encontraron en su estudio de Eumeces egregius, Neoseps reynoldsi y Sceloporus woodi, que los grupos o poblaciones aisladas espacialmente, generalmente presentan profundas divergencias genéticas.
En el presente trabajo se encontraron tres grupos genéticamente diferenciados dentro de los cuales la población de Estación Marte se reconoce como el grupo más alejado, tanto geográfica, como genéticamente, respectivamente, coincidiendo con lo encontrado por Mulvaney et al. (Op. cit.).
Considerando la separación geográfica del grupo de Estación Marte, podríamos inferir un posible aislamiento por distancia, ya que presenta todos los elementos para ello, por lo que se sugiere un análisis de concordancia filogeográfica, mediante la prueba de Mantel para demostrarlo (Wright, 1946; Stalkin, 1994), así como análisis filogenéticos basados en distancias genéticas y caracteres moleculares bajo un modelo evolutivo. Por otro lado, el análisis de la estructura genética poblacional infiere o muestra que el grupo Texas, podría presentar cuatro posibles variedades genéticas. La población de Estación Marte, presentó un haplotipo único, podría representar un posible nuevo grupo genético evolutivamente significativo.
Conclusiones
Uma exsul presentó 22 haplotipos, siendo Bilbao5 el haplotipo ancestral, cuatro de ellos (Estación Marte) candidatos a posibles nuevas unidades evolutivamente significativas. Uma exsul se divide en cinco poblaciones geográficamente diferenciadas y muestra una estructura genética poblacional incipiente e integrada por tres grandes grupos: Bilbao (BAPS1), Texas (BAPS2) y Marte (BAPS3). La población de Estación Marte, por su aislamiento genético y geográfico, se puede considerar una población alopátrica. Según los resultados del análisis mismatch, el cual es unimodal cargado hacia la izquierda, la metapoblación de U. exsul, es una población en expansión, que proviene de un cuello de botella, donde está operando la deriva génica.
Se sugiere el desarrollo de un plan de conservación efectivo para la protección del hábitat que requiere cada grupo genético de U. exsul, con la finalidad de no sólo proteger la especie, que está en estatus vulnerable de conservación, sino conservar la variabilidad genética que permitiría su adaptación a futuras perturbaciones del hábitat.

