











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
La epidermólisis bullosa adquirida (EBA) es una enfermedad ampollosa autoinmune poco frecuente asociada con autoanticuerpos IgG (inmunoglobulina G) que se dirigen contra el dominio NC1 del colágeno tipo VII1, que es el componente principal de las fibrillas de anclaje de la dermis superficial2. La primera descripción de la EBA fue reportada por Elliot en 18951.
Clínicamente, la EBA del adulto es heterogénea, por lo que se han descrito dos formas: la crónica o no inflamatoria y la inflamatoria. La forma crónica se caracteriza por fragilidad de la piel y ampollas "mecánicas" en áreas de traumatismos o fricción, que posteriormente forman cicatrices atróficas y quistes de milium3. La forma inflamatoria imita al penfigoide ampolloso o penfigoide cicatricial. Además, en algunos pacientes se ha descrito una forma mixta4.
La EBA es una enfermedad rara en la infancia; la afectación de mucosas es frecuente y severa5. Los pacientes mejoran con el tratamiento con corticosteroides y sulfonas, aunque los regímenes de tratamiento muestran amplia variación6. Sin embargo, generalmente responden mejor a la terapia convencional y, por lo tanto, presentan un mejor pronóstico que la EBA en adultos.
El objetivo del artículo fue reportar un caso de EBA en una paciente de 12 años.
Caso clínico
Se presenta el caso de un adolescente de 12 años de sexo femenino, quien 13 meses antes de su ingreso a la institución presentó lesiones ampollares en dedos y región periungueal asociadas con prurito, y posteriormente incrementaron en áreas de roce como cuello, dorso, rodillas y dorso de manos y pies.
La paciente no presentó antecedentes personales ni familiares de importancia, con esquema de vacunación completo, y sin hospitalizaciones ni intervenciones quirúrgicas.
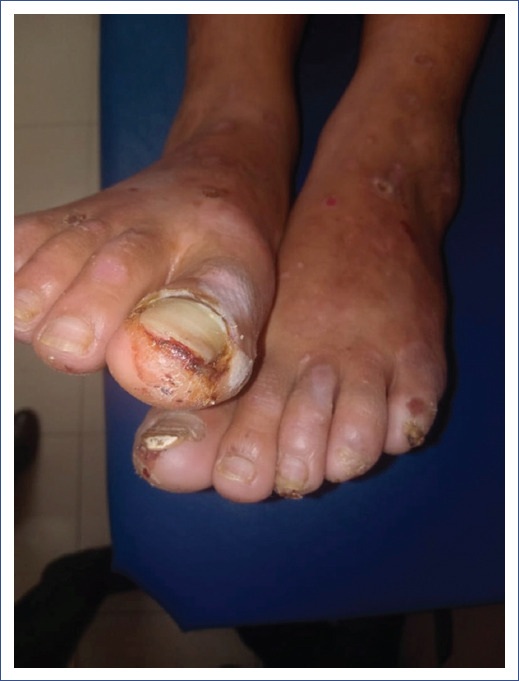
En el examen físico se detectó frecuencia cardiaca de 85 latidos/min; frecuencia respiratoria de 18/min; presión arterial , 110/80 mmHg; temperatura, 36.8°C; peso, 40 kg; talla, 1.51 m. En el examen físico preferencial presentaba leve palidez en piel y mucosas (++/+++); además, se encontraron pápulo-vesículas con algunas ampollas y placas erosivas-costrosas con cicatrización de heridas (que formaban múltiples quistes milium) distribuidas simétrica y bilateralmente en tronco, superficies extensoras de miembros superiores e inferiores (Figura 1). También presentaba distrofia ungueal con degeneración de lecho ungueal en ambos pies (Figura 2). Asimismo, se observó leve compromiso de las mucosas. El resto del examen no mostró alteraciones.

Figura 1 A: pápulo-vesículas con cicatrización de heridas que formaban múltiples quistes milium, distribuidas simétrica y bilateralmente en miembros inferiores. B: presencia de ampollas, pápulo-vesículas y costras con quistes milium, distribuidos en superficies extensoras.
En los exámenes de laboratorio se detectaron los siguientes resultados: hemoglobina: 12.50 g/dl; leucocitos 7.530/mm3; segmentados 4232; linfocitos 2786; plaquetas, 412,000/mm3; proteína C reactiva, 0.08; factor reumatoideo < 7 U/L; glucosa 6 fosfato deshidrogenasa, 13.0 U/g; hormona estimulante de la tiroides (TSH), 2.78; transaminasa glutámico oxalacética (TGO), 14 U/L; transaminasa glutámico pirúvica (TGP), 9 U/L; urea, 22mg/dL; creatinina, 0.47 mg/dL; proteínas totales, 7.2 g/dL; albúmina, 4.8 g/dL; globulinas, 2.4 g/dL; anticuerpos antinucleares (ANA), no reactivo.
Con respecto al estudio histopatológico de la piel perilesional, se encontró ampolla subepidérmica que contenía material fibrinoleucocitario en el piso de ampolla y presencia de edema e infiltrado linfomononuclear, neutrófilos y algunos melanófagos en dermis (Figura 3A). La inmunofluorescencia directa (IFD) reveló depósitos lineales de IgG en la zona de membrana basal, con patrón en U en dermis e IgA, IgM y C3 negativos (Figura 3B). La microscopía electrónica reveló ampolla subepidérmica con membrana basal adherida a la epidermis (Figura 4).

Figura 3 A: estudio histopatológico de piel perilesional. Se observa formación de ampolla subepidérmica que contiene material fibrinoleucocitario en el piso de ampolla. B: estudio de inmunofluorescencia directa de piel perilesional positivo con depósitos de inmunoglobulina G en la membrana basal.

Figura 4 Estudio de microscopia de luz simple. A: ampolla subepidérmica. B: membrana basal adherida a la epidermis. C: piso de la ampolla.
Ante la ausencia de antecedentes familiares de enfermedad ampollar, los hallazgos clínicos, de laboratorio e histopatológicos que mostraron ampolla subepidérmica y la IFD positiva con depósitos IgG en la unión dermoepidérmica permitieron establecer el diagnóstico de epidermólisis bullosa adquirida.
Se administró prednisona a 1 mg/kg/día por 6 semanas y dapsona a 1 mg/kg/día (con el que se encuentra actualmente), ambas por vía oral. La paciente respondió adecuadamente al tratamiento, con disminución de lesiones (Figura 5). En el seguimiento, la paciente continúa con la misma medicación, sin presentar nuevas lesiones en piel.

Discusión
La EBA es una enfermedad ampollosa autoinmune caracterizada por la presencia de anticuerpos anti-colágeno tipo VII, que conduce a la formación de ampollas en la unión dermoepidérmica5. Afecta principalmente a adultos, aunque algunos casos de EBA en niños han sido descritos, sin predilección étnica o de sexo7. La enfermedad es heterogénea con diferentes subtipos: no inflamatorio, inflamatorio y mixto8.
La EBA se caracteriza por el desarrollo de autoanticuerpos contra el colágeno VII, que forma el componente principal de las fibras de anclaje en los hemidesmosomas de la zona densa sublaminar de la piel y las membranas mucosas. La proteína de 290 kDa consta de un dominio de colágeno central flanqueado por dos dominios no colágenos, NC1 y NC2. Los autoanticuerpos dirigidos a dominios NC1 y NC2 se detectan en la EBA. La transferencia pasiva de los anticuerpos contra el colágeno VII activa el sistema del complemento, que libera citocinas inflamatorias y neutrófilos hacia la piel. La formación de complejos inmunes también activa las células natural killer (NK), que atraen a más neutrófilos. Los complejos inmunes se unen al receptor Fc en neutrófilos, lo que lleva a la liberación de especies reactivas de oxígeno y enzimas proteolíticas, que inducen a la formación de ampollas9.
En un estudio de 14 pacientes con EBA infantil, se encontraron cinco pacientes con el tipo inflamatorio y penfigoide ampolloso, y cuatro más con la forma clásica mecanobullosa5. Una revisión de 33 pacientes con EBA infantil también demostró la prevalencia de la EBA inflamatoria, con afectación común de las mucosas6. Independientemente de sus fenotipos, todos los pacientes con EBA presentan autoinmunidad al colágeno tipo VII y la función disminuida de las fibras de anclaje que clínicamente producen fragilidad de la piel, ampollas, erosiones, cicatrices, quistes de milium y pérdida de las uñas10.
Los hallazgos histopatológicos en la EBA incluyen ampollas subepidérmicas con varios grados de infiltrado inflamatorio dérmico4; con la IFD se encuentran depósitos de IgG, C3, IgA e IgM11.
En la actualidad, los hallazgos clínicos, histopatológicos y de estudios de inmunofluorescecia no son suficientes, por lo que Yaoita et al. y Gammon et al.12,13 proponen directrices diagnósticas de EBA: (I) un trastorno ampolloso dentro del espectro clínico definido; (II) falta de antecedentes familiares de un trastorno bulloso; (III) histopatología que revela una ampolla subepidérmica; (IV) IFD de piel perilesional positivo con depósitos de IgG en la membrana basal; (V) microscopía inmunoelectrónica de piel perilesional que indica depósitos de IgG dentro de la lámina densa inferior o la zona densa sublaminar de la membrana basal; y (VI) pruebas de laboratorio alternativo como inmunofluorescencia indirecta.
Uno de los diagnósticos diferenciales más difíciles de excluir es el lupus eritematoso sistémico (LES) ampolloso, donde la ausencia de criterios para esta enfermedad y el aumento de mucina en la dermis reticular pueden ayudar a descartarlo, aunque no es definitivo14. La EBA también se debe diferenciar de otras enfermedades ampollares como el penfigoide ampollar, cuya incidencia en niños es rara y, de presentarse, se caracteriza por ampollas generalizadas predominantes en regiones acrales y rostro, particularmente en niños menores de 1 año, con gran compromiso de mucosas, principalmente la mucosa genital. En el estudio histopatológico típico se evidencia ampolla subepidérmica con eosinófilos; la IFD muestra depósito lineal de C3 en la membrana basal15.
Asimismo, se debe diferenciar entre la EBA y la porfiria cutánea tarda (PCT) infantil, que es poco frecuente y se observa en niños de padres porfíricos, con clínica difícilmente espontánea ya que, por lo general, se expresa como consecuencia de la respuesta del hígado a la agresión de una serie de hepatotoxinas 16. Cabe mencionar que la enfermedad de esta paciente no se asoció con otras enfermedades autoinmunes como LES, artritis reumatoide, enfermedad inflamatoria intestinal y tiroiditis.
Los pacientes con EBA son propensos a complicaciones, como estenosis laríngeas, sinequias laríngeas, simbléfaron y triquiasis y severas deformidades de la mano, incluyendo anoniquia y contracturas.
Según el algoritmo de tratamiento de la EBA propuesto por Ishii et al.10, las formas leves y moderadas de EBA se tratan con corticosteroides sistémicos y colchicina o dapsona como primera opción de terapia adyuvante, mientras que la combinación de corticosteroides sistémicos con agentes inmunosupresores como metotrexato, azatioprina y ciclofosfamida se reserva para el tratamiento de formas intratables de enfermedad.
En el estudio de Delgado et al. en el 2010 se evaluaron pacientes con EBA con seguimiento a largo plazo (hasta 19 años), y se encontró que cuando existe compromiso de la mucosa, puede ser grave, incluso potencialmente mortal.1
En conclusión, la EBA es una manifestación cutánea poco frecuente en niños, así como también su forma no inflamatoria. Es importante reconocer esta patología, ya que puede llevar a formas refractarias o severas, con secuelas como anoniquia o sindactilias y otras que pueden comprometer la calidad de vida del paciente.

