











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
Gorham-Stout disease (GSD), also known as phantom bone disease or vanishing bone disease, is a rare syndrome characterized by lymphatic malformations, mainly in bone structures, that cause progressive osteolysis1. It is more frequent in children and young adults with no observed gender or ethnic preference.
Lymphatic endothelial cell proliferation depends on several growth factors that use the phosphoinositide-3 kinase (PI3K)/Akt pathway, which converges on the mammalian target molecule of the rapamycin (mTOR) pathway. This knowledge has allowed treating GSD with mTOR pathway inhibitors, such as sirolimus or everolimus.
We describe a pediatric patient with rapidly progressing GSD, treated with sirolimus, who demonstrated notorious improvement in ossification and quality of life.
Clinical case
A one-year-old female patient was referred to our institution for diagnosis and treatment. At 8 months of age, after a fall from her stroller, she fractured her right femur and showed a progressive volume increase of the entire right limb and persistent pain. Also, she had a history of ambulatory disability. A computerized tomography scan reported infiltrative blast lesions in both femurs suspicious for neoplasm or Langerhans cell histiocytosis. A positron emission tomography reported destructive lesions of the right femur without regenerative blast activity, demonstrating the partial absence of the proximal third of the right femur. A biopsy of the right femur showed fibroadipose stroma, fragments of immature cartilage with focal ossification, and moderate fibrosis. An inguinal lymph node biopsy showed follicular lymphoid hyperplasia. With this data, the diagnosis of GSD was established. Although the patient was treated with subcutaneous interferon-alpha 2b (IFNa2b) for 18 months, she showed no improvement. She was later referred to our institution’s vascular anomalies department at 3 years of age. She presented with a severe volume increase in her right lower extremity and clinical signs of cellulitis and fasciitis of the thigh, including necrosis and vesicles with hemorrhagic content (Figure 1). The physical examination revealed ulnar clubhand, left tibia varus, and discrete equine left foot. Laboratory tests revealed anemia with hemoglobin of 7.4 g/dL (range: ≥ 11 g/dL), leukopenia of 2.7 x 109 L (range: 5.5 - 15.5 x 109 L), and elevated erythrocyte sedimentation rate at 18 mm/h (range: ≤ 10 mm/h).
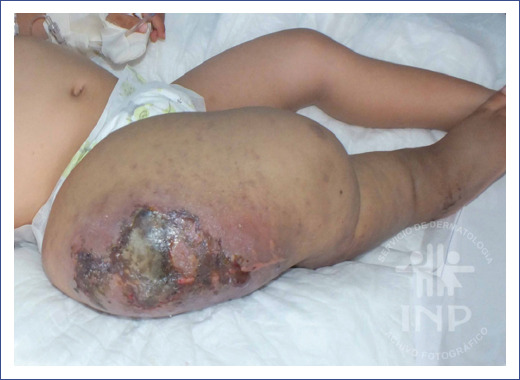
Figure 1 Severe volume increase of the right lower extremity, with clinical signs of cellulitis and fasciitis of the thigh, including areas of necrosis and vesicles with hemorrhagic content.
As the skin culture showed growth of Klebsiella pneumoniae, we initiated treatment with intravenous antibiotics (cefepime 150 mg/kg/day and vancomycin 40 mg/kg/day), resulting in remission of the infection.
Posterior radiological evaluation through serial X-rays and magnetic resonance imaging (MRI) showed lytic lesions in the skull, left femur, both tibias, right radius and ulnae, pseudarthrosis of the left tibia and the right radius, and total absence of the right femur (Figure 2).

Figure 2 A: lytic lesions of the skull. B: lytic lesions in the left femur and tibia, a complete absence of the right femur, and ossification nuclei at the level of the femoral condyles and femoral head are observed. The right hip with loss of acetabulum morphology.
Due to repeated skin and soft tissue infections and progression of osteolysis with null functioning of the right lower extremity at 3-and-half years old, and after cautious debate by the medical team, the right extremity was disarticulated at the hip level. The histopathological report described bone tissue fragments with proliferation of vascular structures with thin wall, flattened endothelium, and dilated congested lumens replacing the medullary canal and causing the neoformation of trabeculae in the bone periphery with prominent osteoblastic rim and marked cortical thinning. Adjacent soft tissues showed fibrosis, vascular engorgement, and foci of recent hemorrhage. Vascular structures were positive for D2-40 staining, confirming GSD’s characteristic lymphatic proliferation.
Treatment with oral sirolimus began at a dose of 0.8 mg/m2/dose every 12 hours, oral vitamin D 800 IU every 24 hours, and intravenous zoledronic acid at a dose of 0.05 mg/kg every 6 months (she received a total of five doses). After 20 months of treatment with sirolimus, the patient reported much less pain, and her laboratory parameters had normalized. Radiological evaluation showed fewer lytic lesions and evidence of ossification in the skull, lumbosacral spine, left femur, left tibia, humeri, radius, and ulnae (Figure 3).

At 8 years of age and after 5 years of treatment with sirolimus, the patient has not presented any infection and only reports mild pain in her left leg and right wrist. There has been a notorious improvement in her quality of life during this time: she now attends school regularly and moves with the support of a skateboard. A prosthesis will be placed as soon as she has more ossification of the lower extremity and spine, with greater load support.
Discussion
Gorham-Stout disease is a very rare disease characterized by intraosseous lymphatic vessel proliferation that causes progressive osteolysis2. The International Society for the Study of Vascular Abnormalities comprises GSD, along with general lymphatic anomalies (GLA) and kaposiform lymphangiomatosis, within the lymphatic malformations3.
GSD mainly affects the epiphysis, most frequently the humerus, femur, and tibia, and less frequently the pelvis, spine, and ribs. It is associated with local deformity, soft tissue enlargement, and considerable pain. In the case of thoracic involvement, chylothorax, and respiratory failure usually develop4. The prognosis is variable, depending on the affected areas and the disease course.
The pathogenesis behind the abnormal proliferation of vascular and lymphatic vessels in GSD remains unknown, although there are different hypotheses considering the vascular endothelial growth factor (VEGF) or the osteoclast hyperactivity. Specific genetic alterations in GSD have not yet been clarified; non-Mendelian mechanisms are proposed, in which a somatic second hit occurs in a causative gene inherited to trigger the lesion5, or it may be caused entirely by pathogenic variants in the KRAS gene6.
Lymphatic and vascular proliferation within the bone suggests that endothelial and lymphatic cells are relevant in stimulating osteoclast differentiation and function by secretion of tumor necrosis factor-alpha (TNFa) and interleukin-6 (IL-6)7, thus generating osteolysis.
GSD diagnosis is based on imaging studies and bone biopsy. In our patient, an MRI demonstrated osteolysis on both femurs (with partial absence of the right), iliac bones, and lumbosacral spine, and hyperintense images in fluid-sensitive sequences as well.
The second biopsy showed the classical findings of GSD: normal bone tissue resorbed and replaced by thin-walled endothelium-lined capillaries of lymphatic origin and D2-40 positive immunostaining that delimits the endothelium of lymphatic vessels8. GLA can also invade bone structures, but cortical bone is not as destroyed as in GSD9.
Sirolimus is an immunosuppressive agent that targets mTOR, specifically, a serine-threonine kinase regulated by PI3K and Akt. The PI3K-Akt-mTOR signaling pathway is closely related to cell growth and proliferation and increases the expression of VEGF, regulating angiogenesis and lymphangiogenesis. As an mTOR inhibitor, sirolimus blocks protein synthesis and has antitumor and antiangiogenic effects7. In lymphatic malformations, sirolimus decreases phosphorylation of PI3K, diminishing the lymphatic endothelial cell proliferation, restoring normal structure in the lesion, and reducing limb volume by correcting lymphatic flow10.
Although there is still no standardized therapy for GSD11, recently sirolimus has been successfully used. In addition to its antiproliferative and antiangiogenic properties12, sirolimus can improve bone homeostasis by inhibiting the mTOR signaling pathway that leads to the differentiation and proliferation of osteoblasts and actively participating in the bone regeneration process13. Ricci et al. studied 18 patients with GSD and GLA that received oral sirolimus and reported an improvement in at least one aspect of the disease in 83% of the subjects14. The most extensive analysis of the use of sirolimus in patients with GSD concluded that the drug appears to stabilize or reduce the signs and symptoms of the disease, improving the quality of life of these patients15.
Among the side effects of sirolimus are diarrhea and stomatitis in schoolchildren, while hypertriglyceridemia has been reported in adolescents16. In our patient, a gradual improvement in quality of life was evident since the first month of treatment with sirolimus: decreased pain, improved mobility, and no adverse effects observed or reported.
In conclusion, GSD is a syndrome characterized by lymphatic malformations causing osteolysis in bone structures and complex management. Treatment with oral sirolimus inhibits osteoclastic activity and angiogenesis, stimulates bone anabolism, and improves ossification, functionality, quality of life, and prognosis in these patients.
Since GSD is a congenital disease with a genetically determined pathophysiology, sirolimus cannot be seen as a curative treatment. However, it should be considered a therapeutic option for decreasing osteolysis progression and improving the patient’s prognosis.

