











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
Duchenne muscular dystrophy (DMD) is an X-linked genetic, degenerative, progressive, recessive inherited disorder caused by mutations in the DMD gene encoding the dystrophin protein1. These patients show metabolic alterations such as hyperinsulinemia, insulin resistance, elevated inflammatory markers, alterations in body composition, and decreased bone mineral density (BMD). DMD is associated with an increased risk of bone fragility2–5 due to the adverse effects of prolonged glucocorticoid therapy and loss of mobility, sometimes resulting in osteopenia and osteoporosis6.
Two processes constantly renew bone: bone modeling and bone remodeling7,8. Both processes involve osteoclastic bone resorption and osteoblastic bone formation. Osteoclasts, multinucleated cells derived from the myeloid hematopoietic lineage, are the primary cells involved in bone resorption, whereas osteoblasts, which carry out bone formation, originate from multipotent mesenchymal stem cells9. Bone remodeling occurs continuously to repair skeletal damage, prevent the accumulation of fragile and hypermineralized bone, and maintain homeostasis. During bone remodeling, osteoclastic resorption is tightly coupled to osteoblastic bone formation. These processes are regulated by the receptor activator of the nuclear factor-kappa B (RANK) signaling pathway, which natural ligands are RANKL and osteoprotegerin (OPG)7-9. As this process is essential for skeletal health, alterations in the RANK/RANKL/OPG complex can disrupt the balance between bone resorption and bone formation, leading to increased bone loss or excessive bone formation9. Increased osteoclastic activity with consequent bone loss has been observed in different pathological conditions, including DMD6,10.
In patients with Duchenne disease, the loss of mobility, together with the administration of systemic corticosteroids, induces a decrease in bone mineral density and favors the development of osteoporosis. This study aimed to highlight some aspects of the RANK/RANKL/OPG system in the remodeling process, the effect of glucocorticoid treatment on this signaling pathway, and the bone health of DMD patients.
Duchenne muscular dystrophy
DMD is the most common neuromuscular childhood disease, with an incidence of one per 3,600 male births worldwide11. DMD is a genetic, progressive, degenerative, X-linked recessive inherited disorder caused by mutations in the DMD gene encoding the dystrophin protein1,12. Dystrophin deficiency in these patients causes cell susceptibility to mechanical stress, eventually leading to abnormal calcium infiltration (a product of sarcolemmal fragility) and interaction with other proteins (calpains, calcium-dependent proteases) that promote muscle fiber destruction13. The mean age at diagnosis is usually 5 years, when the first symptoms appear, including gross motor delay, gait abnormalities (tiptoeing), difficulty rising from the floor using the body to stand up (Gowers’ signs), lordotic posture, and frequent falls due to muscle weakness14. Patients usually lose ambulation and become wheelchair dependent at 10-12 years of age12. In advanced stages of the disease, alterations in pulmonary function due to abnormal development of the thoracic bones (kyphoscoliosis) and weakness of the respiratory muscles (cardiopathy and arrhythmias) are frequent. Cardiorespiratory complications are the most frequent causes of death in these patients15. Patients with DMD present metabolic alterations such as hyperinsulinemia, insulin resistance, elevated inflammation markers, changes in body composition, including BMD2-5, and increased risk of bone fragility due to prolonged glucocorticoid treatment and loss of mobility, sometimes resulting in osteopenia and osteoporosis6.
Diagnosis is indispensable for early intervention. The Multiplex Ligation-Dependent Probe Amplification method is the preferred genetic test for diagnosis, as it analyzes all exons of the DMD gene. However, measurement of serum creatine kinase (CK) concentrations is the most used laboratory test, as CK is elevated in these patients up to 200-fold (1000-3000 IU/L)14. To date, standard treatment in these patients is still based on glucocorticoids. These drugs have been shown to prolong ambulation time by up to 3 years, reduce the deterioration of cardiorespiratory function and the risk of progressive scoliosis, and increase life expectancy12. Current recommendations include initiation of treatment before 5 years of age, as earlier treatment is of more significant benefit regarding loss of ambulation16.
Osteoporosis in DMD
Severe osteoporosis with clinically significant bone fragility has been reported in pediatric patients with DMD17. Currently, osteoporosis is recognized as an essential complication of DMD because it requires follow-up for early diagnosis and interventions to prevent clinically critical sequelae. In this context, the two main risk factors for osteoporosis are progressive muscle weakness with loss of load-bearing capacity and bone toxicity from glucocorticoid therapy18. Clinically, osteoporosis manifests with long bone and vertebral fractures (VF). The latter are frequent, especially in patients treated with glucocorticoids. VFs in the pediatric population with DMD are associated with chronic back pain and spinal deformity, while long bone fractures have been associated with premature and permanent loss of ambulation. Up to 60% of individuals with DMD have lower extremity fractures, often in the distal femur, tibia, and fibula6.
Bone remodeling
The bone system is constantly renewed by two processes: bone modeling and bone remodeling7,8, which involve osteoclastic bone resorption and osteoblastic bone formation. Osteoclasts, multinucleated cells derived from the hematopoietic myeloid lineage, are the primary cells involved in bone resorption, while osteoblasts, which carry out bone formation, originate from multipotent mesenchymal stem cells9. The modeling, resorption, and formation processes occur independently at different sites in the skeleton, producing essential changes in bone architecture. Bone modeling begins in the early stages of skeletal development and modifies the size and shape of the bone. During this process, resorption and bone formation must be uncoupled; bone is removed from one anatomical site, and new bone is formed at another.
An important example of modeling is preserving skeletal shape during linear growth7. Most bone modeling is completed with skeletal maturity, but this process may continue even until adulthood due to an adaptive response to mechanical loading or exercise19. In contrast, bone resorption and bone formation are closely coupled spatially and temporally in remodeling so that bone volume and overall structure remain unchanged. During remodeling, small regions of bone are resorbed by osteoclasts and replaced by osteoblasts; this close coordination between resorption and formation ensures the maintenance of structural integrity and allows for the replacement of up to 10% of the skeleton each year7.
Bone remodeling maintenance requires balancing bone formation, resorption, and direct communication between the different bone cells. Cells of the osteoblast lineage (osteoblasts, osteocytes, and bone lining cells) and bone resorption cells (osteoclasts), together with their precursor cells, are organized into specialized units called bone basic multicellular units (BMUs)20. Several factors control the bone remodeling process. Prominent among these regulatory factors is the RANK/RANKL/OPG system, which sends local and systemic signals that determine the maintenance of equilibrium and the timing of bone resorption and formation within the remodeling cycle7-9.
An increased rate of bone remodeling has been reported in various pathological conditions affecting bone tissue, including postmenopausal osteoporosis, hyperparathyroidism, and rheumatoid arthritis. A decrease in BMD has also been described in different pediatric disorders requiring glucocorticoids, such as leukemias, juvenile rheumatoid arthritis, juvenile systemic lupus erythematosus, asthma, and neuromuscular disorders such as DMD6,10,21.
RANKL/RANK/OPG system
In the late 1980s, understanding of the molecular mechanisms that regulate the formation and activation of osteoclasts advanced rapidly with the discovery of the RANKL/RANK/OPG signaling system, demonstrating that this pathway is key to bone resorption22. This system induces the differentiation and activation of osteoclasts and osteoblasts, balancing the remodeling cycle between formation and resorption. RANKL is secreted by osteoblasts, while RANK, its receptor, is located in pre-osteoclastic cells. The RANKL/RANK interaction induces the formation of multinucleated mature osteoclasts, leading to bone resorption8,9,22,23. A third component, OPG, is also produced by osteoblasts and its binding to RANKL inhibits the RANKL/RANK interaction and thus osteoclastogenesis.
The binding of RANKL to its natural receptor RANK promotes the activation of the intracellular nuclear factor-kB signaling pathway. As a result, the differentiation of preosteoclasts into mature osteoclasts is generated, facilitating bone resorption24. Osteoclast precursors express the macrophage colony-stimulating factor (M-CSF) receptor that promotes the expression of RANK when stimulated. Osteoblasts control osteoclast precursor differentiation by producing RANKL, a cell surface molecule that is the main effector of the RANK receptor, and OPG, a soluble decoy receptor for RANKL22-24.
The binding of RANKL to the extracellular domain of RANK leads to the expression of specific genes involved in osteoclast differentiation, survival, and bone resorption (Figure 1A). The initial step of this signaling pathway is binding TNF receptor-associated factor (TRAF) within the cytoplasmic domain of RANK8,22, leading to NF-kB activation and translocation to the nucleus. TRAFs constitute a family of adaptor proteins, and most of them participate in the activation of the transcription factor NF-kB and the members of the mitogen-activated protein (MAP) family. Different studies suggest that TRAF plays a critical role in the differentiation and activation of osteoclasts8,25-27.
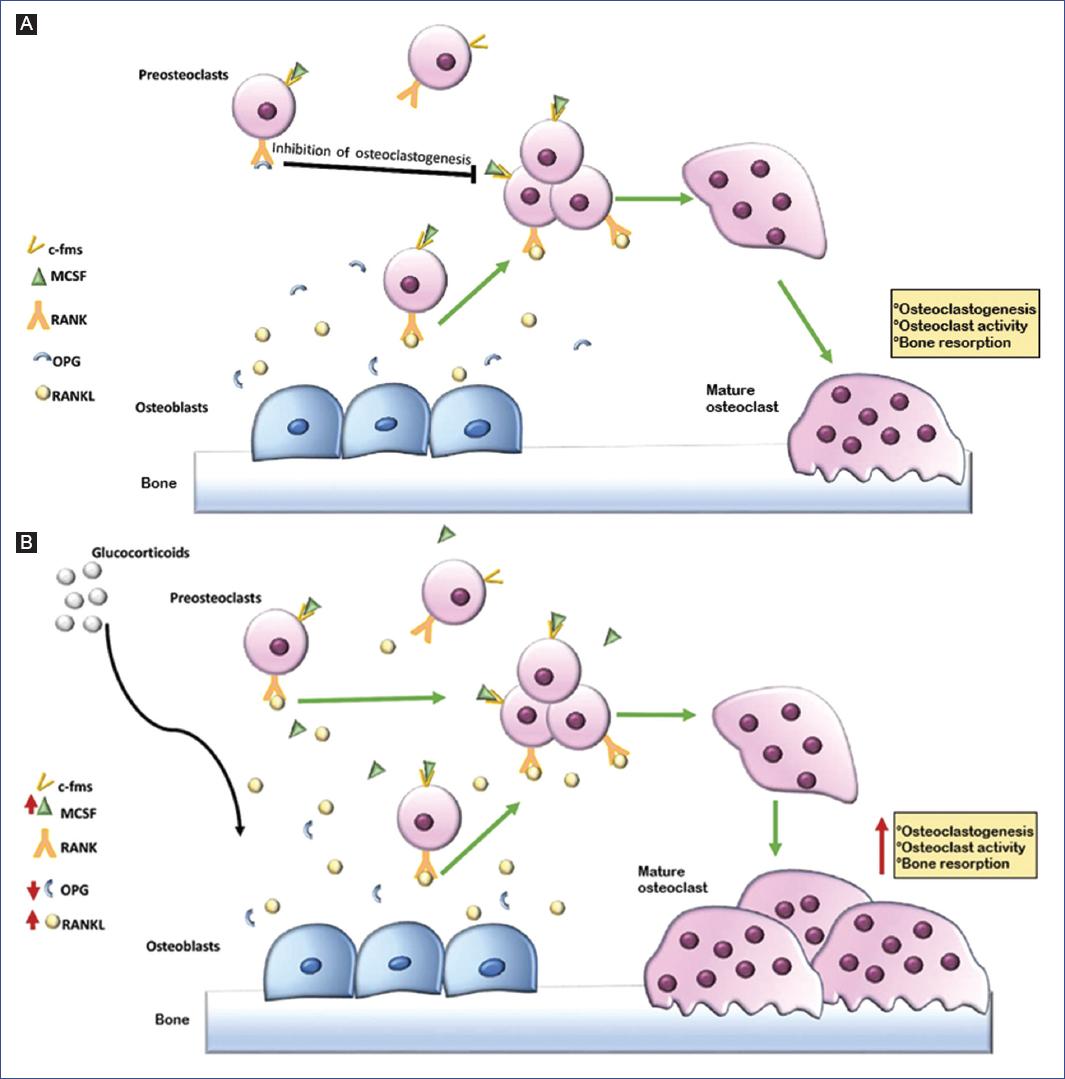
Figure 1 The bone remodeling process is controlled by various factors, among which the RANK/RANKL/OPG system is the most important. This system induces the differentiation and activation of osteoclasts and osteoblasts, balancing the remodeling cycle between bone formation and resorption. A: RANKL is secreted by osteoblasts, whereas RANK, its receptor, is located on preosteoclast cells. The RANKL/RANK interaction induces the formation of multinucleated mature osteoclasts, leading to bone resorption. A third component, OPG, is also produced by osteoblasts and functions as a decoy receptor by blocking the binding of RANKL to its cellular receptor RANK, acting as a negative regulator of RANK signaling, with the ability to inhibit osteoclastogenesis. B: glucocorticoids stimulate RANKL production and OPG inhibition by osteoblastic cells, which promotes osteoclastogenesis and bone resorption. All these factors may contribute to the initial phase of glucocorticoid-induced bone loss. c-fms: colony-stimulating factor-1 receptor; MCSF: macrophage colony-stimulating factor; OPG: osteoprotegerin; RANK: receptor activator of nuclear factor-kappa B; RANKL: RANK natural ligand.
At least seven signaling pathways are activated by RANK-mediated protein kinase: four regulate osteoclastogenesis, and three activate osteoclasts22. OPG acts as a decoy receptor blocking the binding of RANKL to RANK28; thus, it is a negative regulator of RANK signaling, capable of inhibiting osteoclastogenesis25. Osteoblasts also produce OPG in response to anabolic agents such as estrogens and transforming growth factor b (TGF-b), which is related to bone morphogenic proteins (BMPs) and the Wnt/b-catenin pathway28,29. In some studies, osteoporosis has been reported in OPG-deficient mice, while overexpression of OPG or its exogenous administration at high doses induces osteopetrosis-like changes, highlighting the physiological relevance of OPG30-32.
Effect of glucocorticoids on the skeletal system
Glucocorticoids are a group of drugs widely used in different pediatric diseases due to their anti-inflammatory action33. However, their use is associated with numerous adverse effects, including Cushing’s syndrome, diabetes, abdominal obesity, glaucoma, cataracts, growth retardation, hypertension, and musculoskeletal system disorders such as myopathy, osteonecrosis, and osteoporosis34, which are mainly related to high doses and prolonged administration times.
Furthermore, glucocorticoid-induced osteoporosis is currently the leading iatrogenic cause of osteoporosis35, a condition characterized by low BMD and impaired microarchitecture leading to decreased bone strength36. Decreased BMD has been described in different pediatric pathologies with glucocorticoid administration, such as leukemias, juvenile rheumatoid arthritis, chronic renal failure, juvenile systemic lupus erythematosus, asthma, and neuromuscular disorders such as DMD6,10,21.
Glucocorticoid-induced osteoporosis initially presents with an increase in the remodeling rate, a decrease in bone formation, and a reduction in BMPs—this combination of increased bone turnover and negative balance of remodeling results in accelerated bone loss. Subsequently, decreased bone formation predominates, both at the tissue and BMU levels, leading to a state of low bone turnover33.
Glucocorticoids affect bone formation because they inhibit osteoblastogenesis, osteoblasts’ production of bone matrix components, and insulin-like growth factor-I (IGF-I). They also stimulate osteoblast apoptosis and promote osteoclastogenesis by increasing RANKL and decreasing OPG expression in stromal and osteoblastic cells21. Finally, inhibition of bone formation will lead to a decrease in bone remodeling and a sustained increase in the risk of fractures37. These effects are more significant at the trabecular bone level, so changes in these patients are observed mainly in the vertebrae6.
Other mechanisms of glucocorticoids that probably contribute to the decrease in bone tissue are the development of hypogonadism, reduced physical capacity33, inhibition of intestinal calcium absorption, and increased renal calcium losses38. Glucocorticoids have catabolic effects on muscle, in addition to acting on bone and mineral metabolism, producing weakness due to muscle atrophy caused by accelerated muscle protein catabolism. Therefore, glucocorticoid-induced myopathy may contribute to bone deficit21.
Effect of glucocorticoids on RANKL/RANK/OPG system
RANKL and OPG mediate a primary effect of glucocorticoids on the bone system37,39. Glucocorticoids directly stimulate osteoclast differentiation by increasing RANK signaling and indirectly inhibiting interferon-b production. The latter suppresses osteoclast differentiation and stimulates RANKL production and OPG inhibition by osteoblastic cells, thus promoting osteoclastogenesis and bone resorption. These factors can contribute to the initial phase of glucocorticoid-induced bone loss (Figure 1B). When this occurs, patients may present with an impaired state of bone remodeling33,38. Humphrey et al.40 evaluated the effect of different glucocorticoid receptor ligands on OPG and RANKL production and expression in human osteoblastic cell lines, and demonstrated that deflazacort was at least 10-fold less potent as an inhibitor of OPG production compared to prednisolone. Despite this difference in potency, deflazacort was as effective as prednisolone and dexamethasone in inhibiting OPG production at the maximum dose tested (10 µM). Furthermore, deflazacort stimulated RANKL expression by 1.2-fold, whereas prednisolone did so by up to 6-fold.
RANK/RANKL/OPG and Duchenne muscular dystrophy
Muscle hypertrophy or atrophy and gain or loss of bone mineral density occur in parallel in various physiological or pathological conditions. In addition, endocrine, mechanical factors, inflammation, and nutrition can simultaneously affect skeletal muscle and bone metabolism41.
Skeletal muscle and bone form a single functional unit42,43. This muscle-bone unit adapts synchronously during development and periods of modified mechanical loading, such as exercise or prostration or disease (prolonged bed rest, aging, spinal cord injury, critical illness, neuromuscular disease)44-47.
DMD is one of the best examples of synchronization between degeneration, muscle atrophy, and bone loss48,49. Muscle-bone interactions involve bone-derived osteokines, muscle-derived myokines, and dual-origin cytokines that trigger common signaling pathways leading to fibrosis, inflammation, and protein synthesis or degradation. In this regard, myokines can be interleukins, growth factors, or released peptides that can influence remote tissues such as bone. For example, interleukin 6 (IL-6), a proinflammatory cytokine, is among the most studied myokines for its effects on metabolism. IL-6 is a crucial mediator of bone homeostasis and an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Significantly higher IL-6 concentrations have been reported in patients with DMD and mice with X-linked muscular dystrophy (mdx) compared to healthy controls50. Alternatively, blockade of the IL-6 receptor with tocilizumab (a monoclonal antibody directed against the IL-6 receptor) has decreased muscle damage, fibrosis, and regeneration and increased muscle fiber diameter51. Like muscle cells, bone cells release osteokines such as osteopontin (OPN), a known inhibitor of bone mineralization. OPN is also expressed in inflammatory cells, such as macrophages, and its expression is significantly increased during inflammation. OPN is known to promote fibrosis. OPN ablation shifts dystrophic macrophages to a pro-regenerative phenotype, decreasing serum CK concentrations and changes in muscle mass and strength54.
The RANK/RANKL/OPG system regulates bone matrix modeling and remodeling pathways and contributes to muscle pathophysiology in DMD52. Furthermore, RANK, RANKL, and OPG mRNAs are present in skeletal muscle, and RANKL/OPG proteins are found in the myoplasm41,53. Dufresne et al. demonstrated that RANK is expressed in the sarcolemma membrane, potentially interacting with bone-derived RANKL41, and that fully differentiated myotubes secrete OPG, supporting the notion of bidirectional signaling between bone and muscle53. Meanwhile, Zhou et al. compared the expression of bone regulatory myokines in skeletal muscle. These authors compared the mdx model, the dKO model (mice lacking expression of both dystrophin and utrophin, which experience a more severe disease course that phenotypically resembles human DMD), and an age-matched (20-24 weeks) control group (wild-type mice, WT). Among the genes analyzed, the RANKL gene presented a significant increase in mRNA levels in dystrophic muscles, indicating a steady increase in protein concentrations in skeletal muscle tissues of the mdx and dKO dystrophic models compared to WT controls.
Conversely, myostatin showed significant down-regulation in mRNA levels in dystrophic muscles, suggesting a decrease in protein levels in mdx and dKO skeletal muscles compared to WT controls. Regarding the bone phenotypes of the dystrophic mice, computed microtomography analysis of the lumbar vertebrae showed that both mdx and dKO mice significantly decreased bone fraction, number, and thickness of trabecular bone and BMD, and an increase in trabecular spacing compared to WT controls. A significant decrease in mid-femoral bone areas was also observed in dystrophic mice compared to WT controls54.
RANKL participates in different signaling pathways28, including those dependent on NF-kB and Ca2+. Calcium is the primary regulator of multiple intracellular processes in muscle, including myosin-actin cross-linking, protein synthesis and degradation, mitochondrial adaptation, and fiber type switching. The latter is achieved through controlling proteases and transcriptional factors, such as the nuclear factor of activated T cells 1 (NFATc1)55. In osteoclasts, the RANKL/RANK interaction activates Ca2+ and TRAF/NF-kB-dependent signaling pathways, deregulated in DMD52.
A recent study demonstrated that concentrations of several members of the TNF receptor family are significantly elevated in the serum of mdx mice, including the RANK protein, suggesting that it may be involved in the DMD pathophysiology56. To determine the action of the RANK gene in dystrophic skeletal muscle, Dufresne et al. observed a significant improvement in the strength of the long extensor muscles of the fingers in mdx mice with muscle-specific deletion of RANK; however, it had no protective effects against eccentric contraction. These data indicate that the RANK/RANKL/OPG pathway may participate in the pathophysiology of dystrophic muscle.
Furthermore, research in this model has shown that RANK suppression and the use of OPG-Fc mitigate muscular dystrophy by significantly improving muscle strength56. Hamoudi et al. hypothesized that prolonged silencing of the RANKL/RANK signaling pathway would significantly improve DMD. They determined the efficacy of different doses of anti-RANKL in adult mdx mice with utrophin haploinsufficiency (mdx/utrn+/-). The results showed that a 4 mg/kg/3d dose of anti-RANKL significantly improved the specific strength of the dystrophic extensor digitorum longus and soleus muscle compared to mdx/utrn+/- mice treated with phosphate-buffered saline (PBS). However, the treatment did not protect the dystrophic muscles from loss of strength induced by repeated eccentric contraction. Also, hydroxyproline content (bone turnover marker) decreased by approximately 50% after anti-RANKL treatment. Serum CK levels were 6-fold higher in PBS-treated mdx/utrn+/- mice compared to WT mice. Treatment with anti-RANKL significantly reduced serum CK levels by approximately 35% compared with PBS-treated mdx/utrn+/- mice. The authors concluded that RANK/RANKL protein levels are higher in the microenvironment of dystrophic muscles and that inhibition of their interaction with anti-RANKL antibody results in decreased muscle damage, fibrosis, edema, inflammation, and inhibition of NF-kB activity. Anti-RANKL treatment also effectively preserved muscle function and increased bone stiffness in dystrophic mdx/utrn+/- mice57.
Recent studies suggest that circulating levels of OPG could be involved in glucose homeostasis, in addition to being markers of bone metabolism. Bonnet et al. studied the effects of OPG-Fc, considering that RANKL inhibitors could positively influence muscle mass and strength, especially in osteoporosis or sarcopenia, with effects mediated by glucose regulation. The authors evaluated the impact of OPG-Fc administration (4 mg/kg) in muscle for four weeks in the HuRANKLLLTg+ model (characterized by RANKL overexpression) and a Pparb-/- model, which developed a combination of osteo/sarcopenia associated with impaired glucose homeostasis. The authors reported a decrease in muscle mass and strength in the HuRANKLLTg+ group compared to the WT group. In contrast, restoration of cortical bone volume, an increase in limb strength proportional to muscle mass growth, and a marked improvement in insulin sensitivity and muscle glucose uptake was observed in the HuRANKLLLTg+ OPG-Fc group compared to the HuRANKLLLTg+ group, which only received saline. In the Pparb-/- model that received OPG-Fc, an increase in muscle volume and a tendency to gain strength were also observed compared to the control group. The authors concluded that the RANK/RANKL/OPG system plays a crucial role in bone and muscle metabolism58.
As far as we know, few studies have evaluated the RANKL-OPG pathway in pediatric pathologies. Previous reports have shown that blood levels of RANKL are elevated in inflammatory and chronic processes and during treatment with glucocorticoids59.
To date, information on children with DMD is limited to the work of Akhtar et al., who quantified the levels of RANKL, OPG, and the RANKL/OPG ratio in 50 children with DMD and 50 controls (mean age 11.2 ± 0.67 years). In patients with DMD, an inverse relationship between RANKL levels and age was reported (p ≤ 0.0001), whereas controls showed no change (p = 0.52). Alternatively, OPG and the RANKL/OPG ratio also decreased with increasing age (p = 0.026 and p = 0.002, respectively). Seventy-eight percent of glucocorticoid patients tended to have lower RANKL levels than those with no glucocorticoid intake (0.26 pmol/L vs. 0.40 pmol/L, p = 0.05). A possible explanation for these results was that children on glucocorticoid treatment were older (mean age 12.3 years vs. 7.4 years; p < 0.004), which is consistent with the inverse correlation between age and serum RANKL concentrations (rs = -0.6, p ≤ 0.0001). Additionally, RANKL and RANKL/OPG levels were significantly higher in the ambulatory compared to the wheelchair-user group, probably because children with ambulatory DMD were younger (mean age 13.4 years vs. 7.6 years; p < 0.001). However, no correlation was found between RANKL and RANKL/OPG and densitometry Z-index and the presence of fractures. Although the authors inferred that RANK/RANKL/OPG would be involved in the pathophysiology of patients with DMD60, they concluded that some of the bone turnover markers may be difficult to assess or use as therapeutic indicators in this population. More extensive studies, probably cohort studies, are needed to evaluate the role of RANKL-OPG and provide targeted therapy.
Conclusions
Understanding the RANK/RANKL/OPG signaling system has elucidated its importance in the health of the skeletal system and its role in the development of different bone diseases. For example, severe osteoporosis with clinically significant bone fragility has been reported in pediatric patients with DMD. The weakness observed in DMD is partly the result of decreased gait, chronic glucocorticoid use, and changes in the molecular structure of the muscle-bone interaction, highlighting the action of RANK/RANKL/OPG. This system is determinant in maintaining the balance and timing of bone resorption and formation within the remodeling cycle. In addition, the study of the broad biological action of RANKL in other organs and tissues has suggested its activity in the pathophysiology of DMD. As a result, several strategies have focused on using anti-RANKL molecules, RANKL deletion, and OPG, in search of alternatives for treating DMD.

