











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
Las neoplasias de células natural killer (NK) (también conocidas como células asesinas naturales) son entidades poco frecuentes que representan <5% de las neoplasias linfoides y comprometen diferentes entidades clínicas. La Organización Mundial de la Salud, en su clasificación de 2008 de tumores hematopoyéticos y tejidos linfoides, identifica tres enfermedades originadas de las células NK maduras: linfoma extranodal de células NK/T, linfoma nasal de células NK y leucemia agresiva de células NK1. La principal incidencia de esta última se concentra en países de Asia Oriental y América del Sur, donde se ha correlacionado estrechamente con la infección por el virus de Epstein-Barr (VEB).
La leucemia agresiva de células NK fue descrita originalmente por Fernández, et al. (1986) en un paciente de 70 años de edad, con padecimiento de abdomen agudo, que presentó posteriormente síndrome febril, diátesis hemorrágica y muerte2.
Este tipo de leucemia se caracteriza por una proliferación clonal de linfocitos grandes con gránulos CD3- CD56+. Clínicamente, los pacientes presentan principalmente fiebre, además de ictericia, linfadenopatías, hepatoesplenomegalia, células leucémicas en sangre periférica y citopenias. Generalmente, el pronóstico es malo y la mayoría de los pacientes muere en los primeros 2 meses a partir del diagnóstico. Frecuentemente, es posible demostrar la integración de secuencias genómicas del VEB3. Las alteraciones citogenéticas involucradas han sido escasamente caracterizadas: se han reportado deleciones en los cromosomas 6q, 11q, 13q y 17p, asociadas con la iniciación y progresión en el proceso oncogénico4. La enfermedad es típicamente quimiorresistente, lo cual se explica particularmente por la resistencia múltiple a fármacos codificada genéticamente por la glicoproteína P en la membrana celular, que excreta varios agentes citotóxicos como alcaloides de la vinca y antraciclinas. El tratamiento se basa en la administración de metotrexato, etopósido e ifosfamida de inicio, seguido del trasplante alogénico de células progenitoras hematopoyéticas, en caso de lograr la remisión de la enfermedad5.
Caso clínico
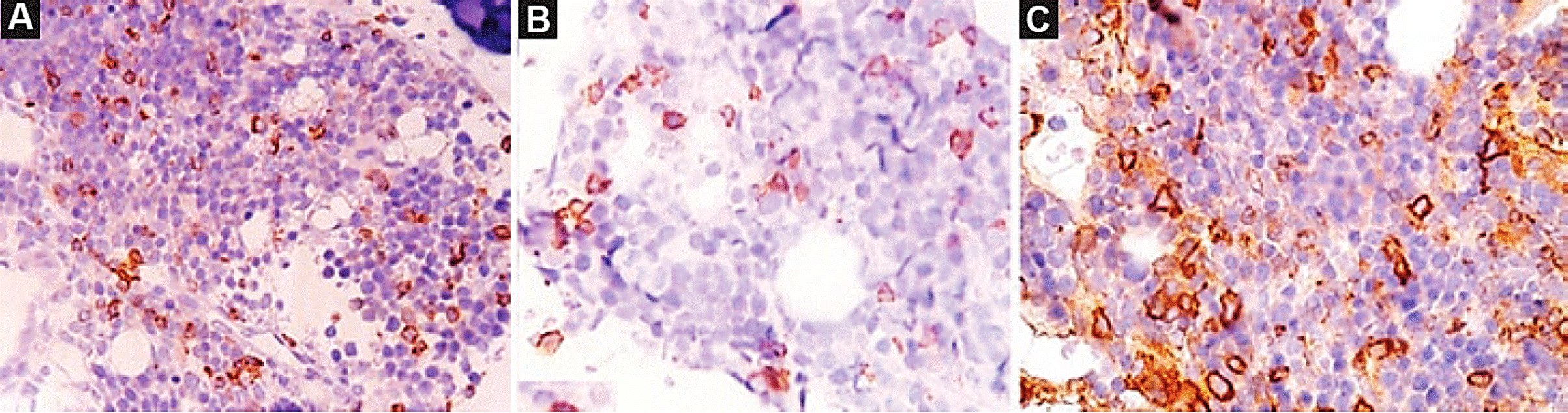
Se trata de un paciente de sexo masculino de 1 año y 7 meses de edad, que inició su padecimiento 3 meses antes de su ingreso en la institución, con presencia de palidez de tegumentos, fiebre intermitente, picos febriles de 38 a 40 °C que duraban 4 horas, dolor óseo, astenia y adinamia. Ante la exploración física, se detectaron adenomegalias submandibulares, soplo ocular, ruidos cardíacos con presencia de soplo sistólico grado II/VI en el segundo espacio intercostal izquierdo y hepatoesplenomegalia. Los resultados de los estudios de laboratorio (biometría hemática) fueron hemoglobina 3.1 g/dl, hematocrito 10%, velocidad corpuscular media 85.3 fl, leucocitos 113,100/mm3, neutrófilos 0%, linfocitos 16%, blastos 84%, plaquetas 224,000/mm3. El resto de los resultados de laboratorio no mostraron alteraciones. Por anemia grave, se realizó transfusión por recambio. Posteriormente, el paciente presentó síndrome de lisis tumoral, y se le implementó manejo basado en soluciones de hiperhidratación (solución glucosada al 5% + bicarbonato de sodio), alopurinol, medidas antihiperkalémicas y controles de laboratorio. En la radiografía de tórax no se observó ensanchamiento mediastinal. Se realizó aspirado de médula ósea, donde se encontró celularidad aumentada, megacariocitos ausentes y blastos de morfología L2 (96%), inmunofenotipo con presencia de CD56 (80.87%), CD34 (35.16%) y desoxinucleotidil transferasa terminal (84.11%) (Fig. 1), índice de DNA de 1.18 y reacción en cadena de la polimerasa con transcriptasa reversa negativa. En el estudio de líquido cefalorraquídeo no se encontraron blastos (SNC 1). Se administró quimioterapia intratecal profiláctica. No se realizó cariotipo en búsqueda de alteraciones cromosómicas, como secuenciación masiva, por falta de recursos y sitio de referencia a nivel nacional para realizar la búsqueda intencionada. En el análisis serológico para el VEB se identificó antígeno (ag) temprano inmunoglobulina (Ig) G (EA-IgG) 14.4 U/ml, ag capsular IgG (VCA-IgG) 650 U/ ml, ag capsular IgM (VCA-IgM) 40.2 U/ml, ag nuclear IgG (EBNA-IgG) 106 U/ml. Se inició tratamiento basado en el protocolo MIED6: altas dosis de metotrexato (5,000 mg/ m2/día, infusión de 24 h i.v. en el día uno), ácido folínico (15 mg/m2/dosis, iniciando a la hora 36 del metotrexato por cinco dosis i.v.), etopósido (200 mg/m2/día en 3 h en los días 2-4 i.v.), ifosfamida (2,000 mg/m2/día en 1 h en los días 2-4) + mesna (500 mg/m2/dosis i.v. cada 6 h), dexametasona (40 mg/m2/dosis cada 8 h por 12 dosis i.v., iniciando en el día uno). El paciente presentó choque séptico posterior al ciclo de quimioterapia. Se realizaron cultivos y se inició terapia con antibióticos intravenosos; no se reportó aislamiento del agente etiológico. Después del primer ciclo de quimioterapia, se realizaron estudios de evaluación, y se documentó remisión clínica y hematológica. La remisión morfológica no fue evaluable, debido a que el aspirado de médula ósea se encontró hipocelular, por lo cual se decidió realizar biopsia de médula ósea. En espera del resultado de patología, se administró metotrexato intramuscular (10 mg/m2/dosis semanal durante cinco semanas)7. Se obtuvo el reporte de la biopsia de médula ósea positiva para infiltración por células neoplásicas con inmunofenotipo CD2+ en el 15%, CD3+ citoplásmico en el 15% y CD56+ en el 25% de la población celular y proteína latente de membrana negativo (Fig. 2). No se encontró genoma de VEB. Se documentó la respuesta parcial con el resultado de la biopsia de médula ósea, pero el paciente presentó nuevamente blastos (células leucémicas) en sangre periférica a las 6 semanas, por lo que se realizó una tomografía de cráneo en busca de linfoma centro nasal, sin hallazgos clínicos. Se decidió iniciar la segunda línea de tratamiento basado en el protocolo SMILE8: altas dosis de metotrexato (5,000 mg/m2/día, infusión de 24 h i.v. en el día uno), ácido folínico (15 mg/m2/dosis i.v. iniciando a la hora 36 del metotrexato por cinco dosis), etopósido (100 mg/m2/día i.v. en 3 h en los días 2-4), ifosfamida (1,500 mg/m2/día en 1 h en los días 2-4) + mesna (500 mg/m2/dosis i. v. cada 6 h), dexametasona (40 mg/m2/dosis i.v. cada 8 h en los días 2-4), L-asparaginasa (6,000 U/m2 en los días 8,10,12,14,16,18 y 20). Este protocolo ha tenido resultados favorables en pacientes con linfoma nasal NK. El paciente presentó toxicidad hematológica posterior al ciclo de segunda línea; persistió con pancitopenia y blastos en sangre periférica. Por presentar respuesta parcial al protocolo MIED, se decidió indicar un segundo ciclo de este, con el objetivo de lograr la remisión para enviar a trasplante de células progenitoras hematopoyéticas haploidéntico (donador materno). Al no lograrse la remisión, se decidió enviar al paciente al programa de cuidados paliativos, pero los padres decidieron buscar una segunda opinión. Hasta el momento, el paciente se encuentra vivo con la enfermedad.

CD3 INT CIT: CD3 citoplásmico; HLA-DR: antígeno leucocitario humano; LLA: leucemia linfoblástica aguda; MPO: mieloperoxidasa; NK: natural killer; TDT: desoxinucleotidil-transferasa terminal.
Figura 1 Inmunofenotipo de la médula ósea.
Discusión
La leucemia agresiva de células NK es una enfermedad rara, con un pronóstico extremadamente sombrío y sobrevida de semanas a pocos meses. Afecta principalmente a personas jóvenes, con predominio del sexo masculino. En este trabajo, se reporta un caso de trastorno linfoproliferativo de células NK, en México, en un paciente menor de 2 años, quien solicitó alta voluntaria al mes del ingreso9. Dada la escasa incidencia de esta variedad de linfomas/leucemias, la información disponible en la literatura actualmente se limita a reportes de casos clínicos y tratamientos empíricos basados en la alta agresividad de estos tumores, a falta de ensayos controlados y aleatorizados.
El padecimiento inicial del paciente simulaba una leucemia linfoblástica aguda, ya que presentó síndrome febril, anémico, infiltrativo e hiperleucocitosis (leucocitos >100,000/mm3), edad de incidencia y presencia de blastos linfoides de morfología L2 (blastos grandes, heterogéneos, cromatina variable, presencia de más de un nucléolo, cantidad de citoplasma moderadamente abundante) en el aspirado de médula ósea. Se detectó inmunofenotipo CD2+, sCD3- (superficie), cCD3+ (citoplásmico), CD56+ y CD16- (este último ausente tanto por inmunofenotipo como inmunohistoquímica), lo cual coincide con lo reportado en la literatura para leucemia agresiva de células NK, por lo que se orientó el diagnóstico con este hallazgo.
Dentro de los diagnósticos diferenciales, se descartó un linfoma centro-nasal de células NK con infiltración a médula ósea mediante una tomografía de cráneo que no presentó alteración. Cabe mencionar que, para confirmar dicho diagnóstico, se debe encontrar una médula ósea con <20% de blastos, característica clínica que el paciente no presentó, confirmándose una leucemia aguda. Estos resultados orientaron al diagnóstico reportado. El caso fue evaluado por la institución de origen, dos instituciones de tercer nivel y una institución a nivel internacional.
El tratamiento descrito en la literatura reporta metotrexato, ifosfamida y etopósido10 como buena terapia inicial, por lo cual se utilizó el protocolo MIED (además de que el paciente presentaba resistencia múltiple a fármacos). Se obtuvo una respuesta parcial (disminución de la cuenta de blastos en la médula ósea >50%, biometría hemática sin citopenias y paciente sin datos clínicos de enfermedad). Posteriormente, se inició la administración de metotrexato intramuscular semanal durante 6 semanas. Sin embargo, el paciente presentó nuevamente actividad de la enfermedad, evidenciada por presencia de blastos en sangre periférica. La eficacia del metotrexato intramuscular se ha documentado en padecimientos crónicos linfoproliferativos benignos al actuar como agente inmunosupresor y no en leucemias agudas como la de este caso.
Se ha documentado la eficacia de la L-asparaginasa11 en estas entidades clínicas, por lo que se decidió utilizar el protocolo SMILE, pues al incluir un fármaco diferente, se podría evadir la resistencia presentada con los otros fármacos. Por esta razón, no se optó por un protocolo de rescate como el IDA-FLAG: fludarabina, citarabina, factor estimulante de colonias de granulocitos e idarrubicina. Se documentó nula actividad y, además, las dosis administradas de los otros fármacos diferentes a la L-asparaginasa eran menores al primer protocolo utilizado.
La enfermedad fue evidenciada nuevamente por la presencia de blastos en sangre periférica y hepatomegalia, por lo que se decidió administrar un segundo ciclo MIED basándose en la respuesta parcial inicial. Existe Bol Med Hosp Infant Mex. 2019;76 controversia en la literatura acerca del uso de antracíclicos11,12, debido a la resistencia que presentan, por lo que no se consideró como opción de tratamiento.
Se han documentado tratamientos exitosos en pacientes con las mismas características que el presente caso con trasplante alogénico o de cordón umbilical. Ebihara, et al. reportaron un protocolo de acondicionamiento con irradiación corporal total (12 Gy en cuatro fracciones por 2 días en los días -9 y -8), tiotepa (150 mg/m2 x 2 en los días -7 y -6) y ciclofosfamida (60 mg/kg x 2 en los días -4 y -3), con presencia de enfermedad y posteriormente trasplante, y tuvieron éxito13. Este protocolo se considera como una posibilidad para este paciente en un tercer nivel de atención. En este caso, el tratamiento definitivo era la realización de un trasplante haploidéntico, con la administración previa de quimioterapia para la lograr la remisión.

