










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkBoletín médico del Hospital Infantil de México
Print version ISSN 1665-1146
Bol. Med. Hosp. Infant. Mex. vol.66 n.5 México Sep./Oct. 2009
Caso clínico
Síndrome de Turner mosaico 45, X/46, XX/47, XXX asociado al síndrome de Klippel-Feil
Turner's syndrome by mosaicism 45, X/46, XX/47, XXX associated to the Klippel-Feil syndrome
Anastasia Morales-Hernández1, Luis Gómez-Valencia1, Miriam Margot Rivera-Angles1, María de los Remedios Briceño-González1, Ezequiel Toledo-Ocampo2, Ramón Miguel Cornelio-García3
1 Servicio de Génetica y Citogénetica
2 Servicio de Pediatría
3 Departamento de Enseñanza, Hospital del Niño Dr. Rodolfo Nieto Padrón, Secretaría de Salud, Tabasco, México
Autor de correspondencia:
Anastasia Morales-Hernández
Correo electrónico: tachimora2_@live.com.mx
Fecha de recepción: 22-05-2008.
Fecha de aprobación: 21-08-2008.
Resumen
Introducción. El síndrome de Turner se debe a la ausencia o anomalía de un cromosoma X, dando como consecuencia talla baja, disgenesia gonadal y estigmas físicos. Se ha descrito su asociación con otras alteraciones como las enfermedades de origen autoinmune y, en raros casos, coexistiendo con el síndrome de Klippel-Feil. Objetivo: informar el caso de una niña con síndrome de Turner por mosaicismo 45, X/46, XX/47, XXX y en la que coexiste el síndrome de Klippel-Feil.
Caso clínico. Paciente femenino con talla baja y estigmas físicos del síndrome de Turner, que presenta limitación para los movimientos de rotación del cuello y adopta una posición obligada de inclinación lateral derecha de su cráneo. El cariotipo mostró un complemento cromosómico 45, X/46, XX/47, XXX; radiológicamente se observó fusión de la primera a la quinta vértebras cervicales y fusión vertebral de la séptima cervical con la primera torácica.
Conclusión. Pudiera representar el primer caso de síndrome de Turner con esta variedad citogenética asociada al síndrome de Klippel-Feil.
Palabras clave. Síndrome de Turner; síndrome de Klippel-Feil; asociación; mosaicismo; síndrome de Turner Mosaico y Klippel-Feil.
Abstract
Introduction. Turner's syndrome is due to the absence or anomaly of an X chromosome, resulting in short stature, gonadal dysgenesis and various physical characteristics. The association of this syndrome with other alterations such as autoimmune diseases has been described and, in rare cases, coexists with Klippel-Feil syndrome. We undertook this study to report the case of a female with Turner's syndrome with mosaicism (45,X/46,XX/ 47,XXX) with the coexistence of Klippel-Feil syndrome.
Case report. We present the case of a female patient with short stature and physical characteristics of Turner's syndrome. The patient presented with limitations of neck movement with a forced position to the right side of her skull. Karyotype showed a chromosomal complement (45,X/46,XX/47,XXX). Radiologically, fusion of the first and fifth cervical vertebrae and vertebral fusion of the seventh cervical vertebra with the first thoracic vertebra were observed.
Conclusion. This may represent the first case of Turner's syndrome associated with a cytogenetic variety of Klippel-Feil syndrome.
Key words. Turner's syndrome; Klippel-Feil syndrome; mosaicim; Turner's syndrome, syndrome Klippel-Feil, association.
Introducción
El síndrome de Turner es originado por ausencia o anomalía estructural de uno de los cromosomas X y se caracteriza fundamentalmente por talla baja, disgenesia gonadal y estigmas físicos.1,2 Turner, en 1938, al estudiar a un grupo de mujeres postpuberales, describió el síndrome que lleva su nombre; caracterizado por talla baja, infantilismo sexual, amenorrea primaria, pterigión colli y cubitus valgus.2 Ortiz y col.3 señalan que fue Ford, en 1959, quien describió por primera vez el cariotipo 45, X en estas pacientes, pero además, estos mismos autores señalaron que hay otras variantes citogenéticas, entre las que se encuentra el mosaicismo XO/ XX. Se ha descrito la asociación de este síndrome con enfermedad inflamatoria intestinal, artritis reumatoide, tiroiditis autoinmune y diabetes mellitus.4
El síndrome de Klippel-Feil se caracteriza clínicamente por cuello corto con limitación de la movilidad e implantación baja de pelo en la región posterior del cuello, debido a la fusión de dos o más vértebras cervicales.5 En la etiología de este síndrome, se han documentado factores ambientales y genéticos que provocan un defecto en el desarrollo embrionario entre las semanas tres y ocho de la gestación, ocasionando falta de segmentación de los somitas cervicales, dando como consecuencia una falta de separación de los cuerpos vertebrales de la columna cervical.6,7 Mackusick5 señala que este síndrome fue descrito por primera vez en 1912 por Maurice Klippel y André Feil, y desde entonces se han documentado asociaciones con otras anormalidades esqueléticas como escoliosis y deformidad de Sprengel, así como hipoacusia, anomalías cardiacas y renales.5,6
En los últimos 30 años se encuentra documentado en revistas indexadas dos trabajos con asociación de los síndromes de Turner y de Klippel-Feil: en 1973 con Hillemand y col.,8 y en 1987 con Suchkova y col.9 Ambas publicaciones se refieren al síndrome de Turner por monosomía X regular (45, X).
El presente trabajo tiene como objetivo informar del caso de una niña con síndrome de Turner por mosaicismo 45, X/46, XX/47, XXX, y en la que coexiste el síndrome de Klippel-Feil.
Presentación del caso clínico
Paciente del sexo femenino de 11 años de edad, originaria y residente de Amatán, Chiapas, México; producto de la primera gesta, de embarazo normo evolutivo (no se refieren antecedentes teratogénicos), de término, parto aparentemente normal, atendido en su domicilio por empírica. Período neonatal sin complicaciones, desconociéndose somatometría al nacimiento. El desarrollo neurológico en su infancia fue retardado: sedestación a los 10 meses de vida, deambulación a los 18 meses, e inició lenguaje con pronunciación de frases a los tres años. Padres no consanguíneos, madre de 16 años de edad y padre de 27 años en el momento del nacimiento de la paciente.
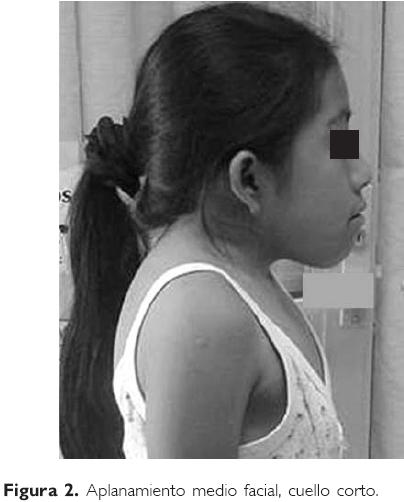
A la exploración física se encontró: peso 27 kg (dentro de la percentil 10), talla 101 cm (por debajo de la percentil 3), por lo que aparenta ser de menor edad, complexión delgada, cráneo con aplanamiento de occipucio, pestañas con alargamiento no usual, fisuras palpebrales horizontalizadas, frente estrecha, puente nasal aplanado, pabellones auriculares de implantación baja, rotados hacia atrás, boca en carpa, paladar alto y ojival, micrognatia. El cuello es corto y ancho, con implantación baja de pelo en la región posterior del mismo y limitación para los movimientos de rotación del cuello. La niña adopta una posición obligada de inclinación lateral derecha de su cráneo (Figs. 1 y 2). Los estudios de rutina consistentes en biometría hemática, química sanguínea y examen general de orina resultaron normales.

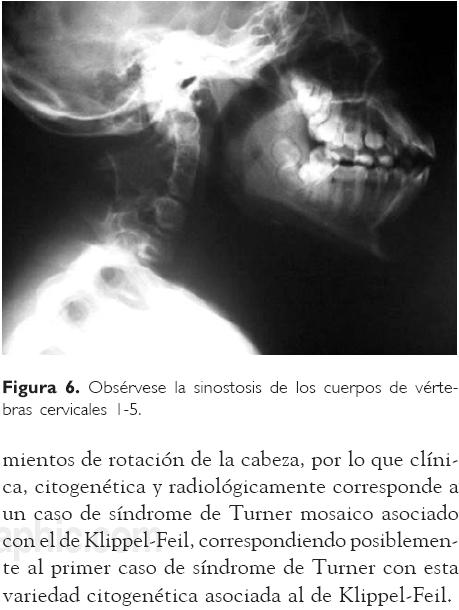
El perfil tiroideo con niveles de T3, T4 y hormona estimulante de tiroides (TSH) normales. El cultivo de linfocitos de sangre periférica para análisis cromosómico evidenció la existencia de tres líneas celulares: 45, X/46, XX/47, XXX, en 64, 24 y 12%, respectivamente (Figs. 3-5)(4). La ecocardiografía mostró normalidad cardiaca, y radiológicamente se observó fusión de la primera a la quinta vértebras cervicales, así como disminución del espacio intervertebral quinto-sexto y sexto-séptimo, además de fusión vertebral de la séptima cervical con la primera torácica (Fig. 6).



Discusión
Uno de cada 400 a 500 recién nacidos vivos presenta anormalidades de los cromosomas sexuales.10 El síndrome de Turner se presenta con una incidencia de 1/2 000 a 1/3 000 niñas recién nacidas vivas y se debe a la ausencia o anomalía de un cromosoma X, dando como consecuencia: talla baja, disgenesia gonadal y estigmas físicos.1 En 50% de los casos hay ausencia de todo un cromosoma X, observándose un complemento cromosómico 45, X, mientras que el otro 50% presenta múltiples anomalías cromosómicas como: mosaicismos, deleciones parciales o translocaciones.11 Dentro de los mosaicismos más comunes se encuentran 45, X /46 , XX; 45, X/46, XX iq; y 45, X/46, XY.11 Se ha descrito la asociación de este síndrome con otras alteraciones como las enfermedades de origen autoinmune, entre las que se incluyen: enfermedad inflamatoria intestinal, artritis reumatoide, tiroiditis autoinmune y diabetes mellitus.4
El fenotipo del síndrome de Klippel-Feil tiene una triada que se presenta en menos de 50% de los casos, caracterizada por implantación baja de cabello, cuello corto y limitación de la movilidad del cuello;12 compartiendo estas características con el síndrome de Turner, con excepción de la limitación de la movilidad del cuello.
En 1973, Hillemand y col.8 describieron el caso de una mujer con síndrome de Turner y en la cual coexistían el síndrome de Rocktansky-Kuster-Hauser y el de Klippel-Feil, y posteriormente, en 1987, Suchkova y col.9 reportaron otro caso de asociación de síndrome de Turner y síndrome de Klippel-Feil; en ambos casos el síndrome de Turner correspondió a la variedad citogenética de monosomía X regular. De igual manera, existen reportes que señalan la asociación del del síndrome de Klippel-Feil con aberraciones cromosómicas de tipo estructural; Clarke RA y col.,13 en 1995, reportaron un tipo de síndrome de Klippel-Feil familiar en el que identificaron una inversión paracéntrica en el brazo largo del cromosoma 8, inv(8) (q22.2 q22.3). Estos autores observaron que los afectados con esta aberración estructural cromosómica presentaban fusión vertebral congénita
El caso que aquí se informa corresponde a una niña de 11 años de edad con un complemento cromosómico que incluye tres líneas celulares: 45, X/ 46, XX/47, XXX, con manifestaciones clínicas de síndrome de Turner y con limitación a los movimientos de rotación de la cabeza, por lo que clínica, citogenética y radiológicamente corresponde a un caso de síndrome de Turner mosaico asociado con el de Klippel-Feil, correspondiendo posiblemente al primer caso de síndrome de Turner con esta variedad citogenética asociada al de Klippel-Feil.
Referencias
1. Heinrich JJ, Martínez A, Pascualini T, Santucci Z, Stivel M. Revisión bibliográfica: Evaluación de tallas finales alcanzadas por pacientes con síndrome de Turner tratadas con hormona de crecimiento. Arch Argent Pediatr. 2001; 99: 239-43. [ Links ]
2. Lozano AEE. Síndrome de Turner: presentación de un caso con menstruación espontánea. Corr Med Cient Holg. 2004; 8: 27-30. [ Links ]
3. Ortiz LC, de Marcos LN, Prieto VM, Farolera BD. Mosaico Turner y embarazo. Presentación de un caso. Rev Cubana Obstet Ginecol. 1998; 24: 24-7. [ Links ]
4. Fernández TT, Espinosa RT, Pérez GC, Pérez SA, García SJ, Carvajal MF. Síndrome de Turner y tiroiditis autoinmune. Rev Cubana Endocrinol. 2003; 14: 13-7. [ Links ]
5. McKusick VA. Klippel Fiel syndrome. Online mendelian inheritance in man. Consultado 26-03-2008. Disponible en: http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=148900 [ Links ]
6. Fernández T, Costa C. Klippel-Feil syndrome with other associated anomalies in a medieval Portuguese skeleton (13th-15th Century). J Anat. 2007; 211: 681-5. Epub 2007; Sept. 7 [ Links ]
7. Klimo P, Rao G, Brockmeyer D. Congenital anomalies of the cervical spine. Neurosurg Clin N Am. 2007; 18: 463-78. [ Links ]
8. Hillemand B, Bonneau JC, Joly JR. Conjunction of Turner's syndrome, atypical Rokitansky-Kuster-Hauser syndrome and Klippel-Feil syndrome. Ann Med Intern (Paris). 1973; 124: 423-8. [ Links ]
9. Suchkova EN, Panova TN, Egorova SP, Poliakova GA. Shereshevskii-Turner syndrome in a patient with Klippel-Feil syndrome. Klin Med (Mosk). 1987: 65: 127-8. [ Links ]
10. Ramírez GC, Herrera JC, Durango NE, Ramírez JL. Cariotipo 45, X, r(X) en pacientes con diagnóstico clínico de Turner. IATREIA. 2000; 13: 161-6. [ Links ]
11. Velasco H, García N, Madero JI. Complicaciones materno-fetales en pacientes con síndrome de Turner. Reporte de dos casos manejados con donación de óvulos. Rev Colomb Obstet Ginecol. 2006; 57: 117-23. [ Links ]
12. Austrich SE, Téllez ZJ, García RG, Corona R. Síndrome de Klippel-Feil. Imágenes por tomografía en tercera dimensión. Gac Med Mex. 2001; 137: 609-12. [ Links ]
13. Clarke RA, Singh S, Mckenzie H, Kearsley JH, Yip MY. Familial Klippel-Feil syndrome and paracentric inversion inv(8) (q22.2 q22.3). Am J Hum Genet. 1995; 57: 1364-70. [ Links ]

