











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkAortic coarctation accounts for 5-8% of congenital heart defects. When accompanied by other congenital heart defects, it is called complex aortic coarctation. Its association with bicuspid aortic valve is between 60% and 85%.1 Without correction of the defect, mean survival of these patients is 35 years. Survival greater than 65 years is extremely rare and few cases have been reported in the literature.2
We report the case of a 68 year-old woman with history of resistant hypertension since early adulthood who presented progressive dyspnea during the last two years.
Physical exploration was remarkable for short stature, underdevelopment of lower extremities and laterally displaced apical impulse. Auscultation over the aortic area revealed a harsh meso-telesystolic crescendo-decrescendo murmur IV/VI radiating to carotids and apex with Gallavardin phenomenon. A loud systolic murmur was also heard over the left paravertebral area. Carotid pulse was parvus et tardus. Lower extremities pulses where markedly diminished. Blood pressure was 152/90 mmHg in the upper extremities and 64/40 mmHg in the lower extremities. Ankle brachial index was 0.42. Chest radiograph showed grade III cardiomegaly, radiographic data compatible with the ‘‘3’’ sign, inferior rib notching (Roesler sign) and calcified collateral vessels (Fig. 1A). The EKG demonstrated a sinus rhythm, complete right bundle branch block, left anterior fascicular block, biventricular hypertrophy and systolic overload of the left ventricle (Fig. 1B). Transthoracic echocardiography showed a calcific severely aortic stenosis with a mean gradient of 44 mmHg, peak gradient of 73 mmHg and a peak velocity of 4.2 m/s. Left ventricular ejection fraction was 49%, there were no regional wall motion abnormalities. Transesophageal echocardiography confirmed a stenosed, functionally bicuspid aortic valve, aortic valve area was 0.6 cm2 by planimetry (Fig. 2A and B). CT angiography revealed a markedly diminished descending aorta with a minimum diameter of 4 mm measured 35 mm after the origin of the left subclavian artery (Fig. 2C and D). Aortic root and ascending aorta size were normal (26 and 36 mm respectively).
Left heart catheterization revealed a trans-coarctation peak to peak pressure gradient of 110 mmHg, pre-coarctation pressure was 200/63 mmHg and post-coarctation pressure was 90/54 mmHg. The trans-aortic peak to peak pressure gradient was 28 mmHg, left ventricular pressure was 226/4 mmHg and ascending aorta pressure
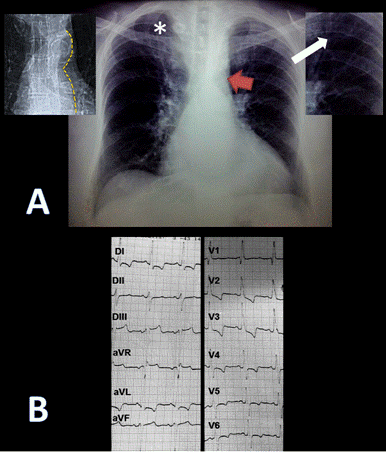
Fig. 1 (A) Posteroanterior chest radiograph demonstrating situs solitus, levocardia, levoapex, left sided aortic arch, normal pulmonary blood flow and grade III cardiomegaly, additionally the ‘‘3’’ sign (red arrow), inferior rib notching (white arrow) and calcified collateral vessels (*). Top left panel delineating the descending aorta contour from the same patient in the thoracic scout CT image. Top right panel is a zoom of the inferior rib notching. (B) Standard 12 lead EKG demonstrating a sinus rhythm, complete right bundle branch block, left anterior fascicular block, biventricular hypertrophy and systolic overload of the left ventricle.
was 198/60 mmHg. No significant coronary artery stenosis were found.
Based on the association of a bicuspid aortic valve and a coarctation of the aorta, she was diagnosed with a complex aortic coarctation and severe aortic valve stenosis. Considering the anatomic complexity for an endovascular treatment (Fig. 2E), the patient was proposed for a surgical coartectomy and aortic valve replacement in a two stage surgery approach. The heart team accepted the surgical proposition but the patient refused the surgical intervention during the index hospitalization. She continues the follow up while considering the intervention. Notably, without a surgical correction, the survival likelihood of this patient is poor in the middle term, the mortality of patients with severe aortic stenosis presenting with heart failure is around 50% at 2 years, furthermore, the added risk of aortic rupture/dissection and cerebral hemorrhage in patients with aortic coarctation confers a bad prognosis.
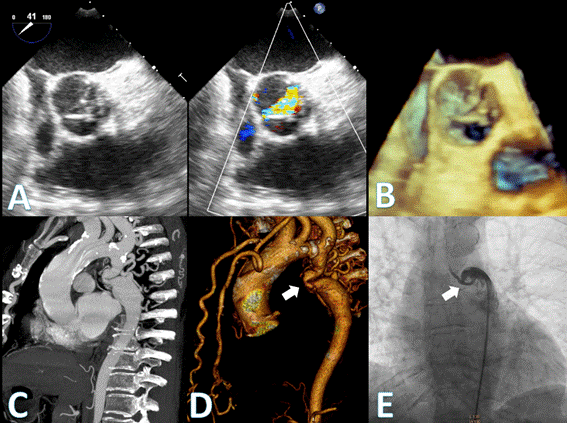
Fig. 2 (A) Transesophageal echocardiography aortic short axis view showing a Type 1 bicuspid aortic valve (right-left coronary cusp fusion) with important thickening and color Doppler acceleration. (B) 3D imaging of the aortic valve. (C) Maximum intensity projection CT angiography and (D) volumetric reconstruction showing the severe aortic coarctation site (white arrow) measured 35 mm after the origin of the left subclavian artery along with marked collateral circulation (mammary and vertebral arteries). (E) Coarctation site angiography demonstrating the reduction of the luminal area (white arrow) and the anatomic complexity for an endovascular treatment.

