










Services on Demand
Journal

Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
 Cited by SciELO
Cited by SciELO Related links
 Similars in
SciELO
Similars in
SciELO Share

 Permalink
PermalinkArchivos de cardiología de México
On-line version ISSN 1665-1731Print version ISSN 1405-9940
Arch. Cardiol. Méx. vol.81 n.1 Ciudad de México Jan./Mar. 2011
Artículo de revisión
Myocardial postconditioning: anesthetic considerations
Posacondicionamiento miocárdico; consideraciones anestésicas
Pastor Luna–Ortiz,1 Juan Carlos Torres,1 Gustavo Pastelin,1 Martín Martínez–Rosas2
1 Department of Pharmacology
2Department of Physiology
3 Instituto Nacional de Cardiología Ignacio Chávez, México, D.F.
Corresponding author:
Martín Martinez, Department of Physiology.
Instituto Nacional de Cardiología Ignacio Chávez,
México, DF. Juan Badiano No. 1. Colonia Sección 16,
México, DF. CP 14080.
E–mail: martin5163@yahoo.com
Recibido el 12 de marzo de 2010;
Aceptado el 12 de enero de 2011.
Abstract
Recently, it has been shown that the heart can be protected against the ischemia–reperfusion injury if brief coronary occlusions are performed just at the beginning of the reperfusion. This procedure has been called postconditioning (PostC). It can also be elicited by pharmacologicalinterventions, which are named pharmacological PostC. In general, PostC reduces the reperfusion–induced injury, blunts oxidant–mediated damages and attenuates the local inflammatory response to reperfusion, decreases infarct size, diminishes apoptosis, neutrophil activation, and endothelial dysfunction. The mechanisms that participate in PostC are still not completely understood. In this regard, adenosine, glycine, bradykinin, ciclosporin A are involved in PostC triggering. Similar to ischemic preconditioning, PostC triggers several signaling pathways and molecular components, including nitric oxide (NO), protein kinase C, adenosine triphosphate–sensitive potassium channels, the Reperfusion Injury Salvage Kinases (RISK) pathway, which comprises phosphatidylinositol–3–OH kinase (PI3K) and extracellular signal–regulated kinase (ERK 1/2), and, finally, the Survivor Activating Factor Enhancement (SAFE) pathway. In this review, we describe the mechanisms of reperfusion–induced injury as well as the proposed protective pathways activated by PostC, which seem to converge in inhibition of mitochondrial permeability transition pores opening. On the other hand, experimental evidence indicates that volatile anesthetics and opioids are capable of exerting cardioprotective effects under certain conditions, constituting a very useful pharmacological PostC. Thus, the first minutes of reperfusion represent a window of opportunity for triggering the aforementioned mediators, which acting in concert lead to protection of the myocardium against reperfusion injury. Pharmacological, especially anesthetic, PostC may have a promising future in the clinical scenarios in the operating room.
Keywords: Postconditioning; cardioprotection and isquemia–reperfusion injury; Mexico.
Resumen
Recientemente, se ha demostrado que el corazón puede protegerse contra el daño por isquemia–reperfusión si se aplican breves oclusiones coronarias justo al inicio de la reperfusión. Este procedimiento ha sido llamado posacondicionamiento y puede ser producido mediante intervenciones farmacológicas, las cuales constituyen el posacondicionamiento farmacológico. En general, el posacondicionamiento reduce el daño inducido por la reperfusión, disminuyendo el daño oxidativo y atenuando la respuesta inflamatoria local durante la reperfusión, así también disminuye el tamaño del infarto, disminuyendo el proceso de apoptosis, la activación neutrofílica y la disfunción endotelial. Los mecanismos que participan en el posacondicionamiento aún no son bien entendidos, aunque se sabe que moléculas como la adenosina, la glicina, la bradicinina y la ciclosporina A están involucradas en la activación del posacondicionamiento. De manera similar al preacondicionamiento isquémico, el posacondicionamiento activa rutas de señalización en las cuales participan diversos componentes moleculares como el óxido nítrico, la proteína cinasa C, los canales sensibles a ATP, la ruta de aumento del factor de activación de sobrevivencia, así como la ruta de las cinasas de salvamento de la lesión por reperfusión las cuales comprenden la cinasa de fosfatidilinositol–3–0H y la cinasa regulada por señales extracelulares. En esta revisión describimos los mecanismos de daño inducido por la reperfusión así como las vías protectoras propuestas activadas por el posacondicionamiento, las cuales parecen converger en una inhibición de la apertura de los poros de transición de la permeabilidad mitocondrial. Por otro lado, la evidencia experimental indica que los anestésicos volátiles y los opiáceos son capaces de ejercer efectos cardioprotectores bajo ciertas condiciones, constituyendo un posacondicionamiento farmacológico muy útil. De esta manera, los primeros minutos de la reperfusión representan una ventana de oportunidad para activar los mediadores antes mencionados, los cuales actúan en concierto para llevar a la protección del miocardio contra el daño por reperfusión. El posacondicionamiento farmacológico especialmente el anestésico puede tener un futuro promisorio en los escenarios clínicos de las salas de operaciones.
Palabras clave: Posacondicionamiento; cardioprotección y daño por isquemia–reperfusión; México.
Introduction
Despite advances in prevention and treatment, cardiovascular disease remains as the number one cause of death in men and women in the United States.1 Acute myocardial infarction (AMI) is still a frequent and disabling disease being the infarct size a major determinant of myocardial functional recovery and mortality after an AMI.2 Thus, limitation of infarct size represents an appropriate strategy to prevent postinfarction heart failure and to improve survival. In the last 15 years, there has been a significant improvement in the outcome of AMI patients. This improvement is mainly based on the introduction of efficient reperfusion strategies, such as primary percutaneous coronary intervention (PCI) and thrombolysis.3 However, coronary heart disease still presents a mortality rate of around 10% at one year for all AMI patients.1 This remaining high mortality rate is partly a consequence of the phenomenon called "myocardial reperfusion injury",4 originally described by Jennings and Sommers5 in the sixties in an experimental model of myocardial infarction. In the myocardial reperfusion injury, the reperfusion causes additional functional and structural damage to the myocardium after a period of ischemia when the myocardium is acutely reperfused.5 Studies of AMI in animal models suggest that lethal reperfusion injury, which starts immediately after the opening of the culprit coronary artery, accounts for up to 50% of the final size of a myocardial infarct.4 In human clinical studies, this phenomenon is associated with detrimental myocardial remodeling, ventricular arrhythmias, and no–reflow, and predicts a negative prognosis in myocardial infarction patients.6 Thus, lethal reperfusion injury has become a major therapeutic target in the search for improvement in AMI patient's recovery and numerous strategies have been tested in experimental settings to reduce reperfusion injury. Recently, it has been shown that the heart can be effectively protected from reperfusion injury not only with ischemic preconditioning (IPC), which is a classic strategy of cardioprotection, but also with brief episodes of ischemia during the early reperfusion period after a prolonged period of ischemia. This process is called: ischemic PostC.
In this review, we describe the mechanisms of reperfusion injury, the possible protective mechanisms of the PostC and pharmacological PostC, especially using anesthetics to induce the heart protection.
Mechanisms of reperfusion injury
The heart is a pump that converts chemical energy into mechanical work. The power produced by the cardiac muscle is generated almost entirely by oxidation of carbon fuels with oxygen. The fuels are provided to a great extent by coronary blood flow. Oxidative metabolism is the function of mitochondria within the cell, and thus the bulk of cardiac energy is supplied by oxidative phosphorylation within cardiac mitochondria. Like many cells, when deprived of oxygen (anoxia), cardiac cells can maintain ATP levels by anaerobic glycolysis, and can then revert smoothly to oxidative metabolism on reperfusion.7 However, if blood flow is restricted, as in myocardial infarct, the cells accumulate glycolytic by–products (lactate, H+) in addition to suffering from oxygen deprivation.8 This is a condition known as ischemia and can damage cardiac cells irreversibly. Paradoxically, however, the major damage to ischemic cells comes on the re–introduction of oxygen (reperfusion). During reperfusion, the cells typically undergo further contraction (hypercontracture) and membrane damage, followed by cell death.9,10 The reperfusion injury of the myocardium is a complex phenomenon that encompasses a list of events including: reperfusion arrhythmias, microvascular damage, reversible myocardial mechanical dysfunction ("stunned" myocardium), and cell death (due to apoptosis or necrosis). These events may occur together or separately. Most of the detrimental effects of reperfusion are triggered within the first minutes following the re–opening of the occluded coronary artery.10 However, most of the cellular disturbances that occur at the time of reperfusion are determined by the abnormalities induced during the ischemic period (Figure 1). During ischemia, the increase in anaerobic glycolysis results in the progressive accumulation of protons and lactic acid, with increased acidosis eventually inhibiting glycolytic flux and synthesis of ATP. As the cardiomyocyte attempts to correct acidosis through the Na+/H+ exchanger (this transporter moves H+ out of the cell and Na+ into the cell), the Na+ is accumulated and this event activates the Na+/K+ ATPase to pump Na+ out of the cell. However, taking in account the diminished ATP production, the Na+/K+ ATPase progressively fails without sufficient ATP. Subsequent activation of the Na+/ Ca2+ exchanger, in its reverse mode, helps to move Na+ out of the cell, but favors cytosolic accumulation of Ca2+. Prolonged ischemia induces a progressive failure of ionic homeostasis, which ultimately causes accumulation of intracellular Na+ and Ca2+ and a decline in ATP levels. If ischemia continues for a long time, a hypercontracture is developed and cell death occurs.
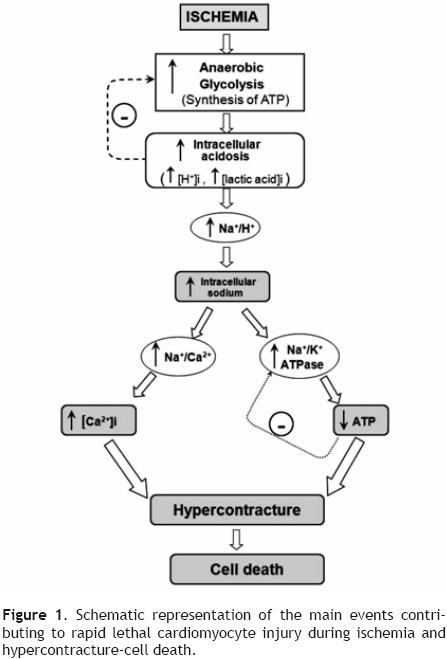
During early reperfusion (in the first minutes), occurs a rapid correction of acidosis (low intracellular pH) through the Na+/H+ exchanger, and through the entry of the ion HCO3– by the Na/HCO3 symporter (Figure 2). Additionally, the lactate, previously accumulated, is washed out with the restored blood flow diminishing the acidosis. However, all these events cause accumulation of Na+. This additional augment in intracellular sodium induces a secondary activation of the Na+/Ca2+ exchanger in the reverse mode to move out Na+ ions; however, this aggravates cytosolic Ca2+ accumulation. On the other hand, the abrupt re–exposure of the ischemia–inhibited mitochondrial respiratory chain to oxygen generates a membrane potential to drive ATP synthesis, which leads to a rapid overload of Ca2+ in the matrix and massive production of reactive oxygen species (ROS), which by themselves are capable of damaging cellular membranes and to induce oxidative stress. These two factors (cytosolic Ca accumulation and oxidative stress) trigger the mPTP to open, and this pore is a key element in cell death.11 The mPTP is a nonspecific channel located on the inner mitochondrial membrane. The mPTP was originally proposed to consist of three major components: a voltage–dependent anion channel, the adenine nucleotide translocase, and cyclophilin D, contained within the mitochondrial matrix, however, its structure is not completely defined. Under normal physiological conditions, the mitochondrial inner membrane is impermeable to almost all metabolites and ions, and the mPTP is in a closed conformation. Under some stress conditions, the mPTP may open and trigger several deleterious events. Opening of the mPTP abolishes the mitochondrial membrane potential (Δψm) inhibiting oxidative phosphorylation. The mPTP opening allows the equilibration of molecules that are smaller than approximately 1500 daltons. The osmotic force generated by matrix proteins results in matrix swelling (mitochondrial swelling), leading to further rupture of the outer membrane and to the release of proapoptotic factors, such as cytochrome c, into the cytosol. These actions rapidly produce cell death. In addition, disruption of the mitochondrial membrane potential also causes the ATP synthase to behave as an ATPase and accelerates energy depletion secondary to the ischemic insult.12
Experimental evidence supports that mPTP opening is a postischemic event. In the isolated rat heart model, it has been demonstrated that the cytosolic release of NAD+ (this molecule was used as a marker of mPTP opening) occurs at the time of reperfusion following a prolonged ischemic insult.13 Griffiths and Halestrap used the [3H]2–deoxyglucose entrapment technique to investigate the kinetics of in situ mPTP opening, and demonstrated that mPTP opening does not happen during ischemia, but occurs within the first 5 minutes of reflow following a 30–minute period of ischemia in the isolated rat heart.14 Importantly, the time course of mPTP opening appeared to match the rapid correction of pH that occurs at reperfusion. Recent in vivo studies support this concept by showing that PostC may mediate its cardioprotective effects through prolonged transient acidosis during the early reperfusion phase.15
Inflammatory changes and endothelial function during reperfusion
During reperfusion there are several altered responses in other tissues or systems besides the cardiac muscle forming a set of events that make up the reperfusion damage. Alterations of endothelial function are pivotal in the development of reperfusion damage and the no–reflow phenomenon. The inflammatory process, characterizing early and late periods of reperfusion, is an important aspect of changes leading to tissue damage by its effects on endothelial cells. Neutrophils feature prominently in the inflammatory component of postischemic injury. This occurs because ischemia–reperfusion prompts a release of oxygen free radicals, cytokines, and other pro–inflammatory mediators that activate both the neutrophils and the coronary vascular endothelium.16 Activation of these cells promotes the expression of adhesion molecules on both neutrophils and the endothelium, which recruit neutrophils on the endothelial surface and initiate a specific cascade of cell–cell interaction. This leads first to the adhesion of neutrophils to the vascular endothelium and subsequently to their transendothelial migration and their direct interactions with the interstitial matrix and myocytes.17,18 This specific series of events is a prerequisite for the full expression of reperfusion injury, including endothelial dysfunction, microvascular collapse, and impairment of blood flow (the "no reflow" phenomenon), myocardial infarction and apoptosis.
Postconditioning to protect the heart
In the procedure called myocardial PostC, the heart can be protected against the ischemia–reperfusion injury with brief coronary occlusions performed just at the beginning of the reperfusion. PostC was first described by Zhao and colleagues in dogs,19 in which it reduced the myocardial injury to an extent comparable to IPC. Beneficial outcomes observed with PostC include reduction in infarct size,20,21 in endothelial dysfunction, in neutrophil adherence,19,21 and in apoptosis.22 Recently, it has been shown that a range of pharmacological agents given at the moment of reperfusion after an ischemic insult can significantly protect the myocardium, and this effect has been shown to involve protection against necrosis and apoptosis. This maneuver has been called: pharmacological PostC.
Proposed mechanisms of PostC
Studies have documented that several pathways and molecular components are involved in the cardioprotective effects of PostC. These include NO,23 phosphatidylinositol 3–kinase (PI3K),24 extracellular signal–regulated kinase (ERK),25 PKC,26 and mitochondrial adenosine triphosphate–sensitive potassium channels (mitoKATP),27 Reperfusion Injury Salvage Kinases (RISK), and Survivor Activating Factor Enhancement (SAFE) pathways. However, there is not a scheme that could help to understand the role of each described component and the final or direct effectors of PostC.
mPTP as potential end–effector of PostC
As described, mitochondria and mPTP opening have been proposed to play an essential role in reperfusion injury28 and, in this way, inhibition of mPTP opening has been reported to be an important mechanism underlying PostC protection.29 A number of studies have discovered a common finding with regard to the timing of PostC. The protection induced can only be taken advantage if PostC is initiated at the onset of reperfusion and is lost if it is delayed by few minutes in rat30 and rabbit models.31 Therefore, this would suggest that the end–effector of protection must exert its actions during the initial stages of reperfusion. In this regard, pharmacological inhibition of the mPTP during the early minutes of reperfusion32 has been shown to be cardioprotective, however delaying this inhibition by 15 minutes abolished this protection. Similarly, delaying the administration of insulin, which is known to activate the RISK pathway, until after the first 15 minutes of reperfusion abrogates its infarct–limiting effect.33 Therefore, one could speculate that the mPTP, which is known to regulate cell death during the first few minutes of reperfusion,34 is potentially the main candidate as the end–effector. Other studies have assessed the interaction between the mPTP opening, and the postconditioning interventions. In a rabbit model of ischemia–reperfusion, Argaud and colleagues found that both, mechanical (initiating with a smooth flow at the beginning of reperfusion) and pharmacological PostC with cyclosporin A (CsA) or NIM811, a specific inhibitor of the mPTP, that was given 1 minute before reperfusion, limit infarct size by at least 45% compared with control animals.35 Thus, they demonstrated that the suppression of mPTP opening at reperfusion provided powerful antinecrotic and anti–apoptotic protection to the ischemic myocardium. They also showed that both mechanical and pharmacological PostC increase the Ca2+ load that is required to open the mPTP.36 Additional evidence for a major role of the mPTP in lethal reperfusion injury recently came from the use of transgenic mice lacking cyclophilin D (CypD).37,38 CypD, which is recognized as a key molecular component of the mPTP, is a mitochondrial member of the family of peptidyl–prolyl cis–trans isomerases (PPIases). Although still debated, it was reported that, in the presence of a high matrix Ca2+ concentration, CypD modifies the conformation of the inner membrane proteins to form a mega–channel. The molecular structure of the mPTP remains poorly known and, besides, CypD might involve several other proteins including voltage–dependent anion channel (VDAC) or adenine nucleotide translocator (ANT), as mentioned above. Unfortunately, their precise role is still elusive and no pharmacological agent targeting these proteins is currently available for clinical trials. In vivo, CypD–deficient mice develop smaller infarcts after a prolonged coronary artery occlusion followed by reperfusion.37, 38 Recently, Lim et al. reported that CypD–deficient mice cannot be postconditioned, further suggesting that lethal reperfusion injury is mediated by mPTP opening.39 These results strongly support the proposal that mPTP opening, triggered by mitochondrial Ca2+ overload and overproduction of ROS, plays a central role in lethal reperfusion injury, and specific inhibition of mPTP opening at the time of reperfusion is central to the cardioprotec–tive effect induced by PostC.
Additional mechanisms of PostC
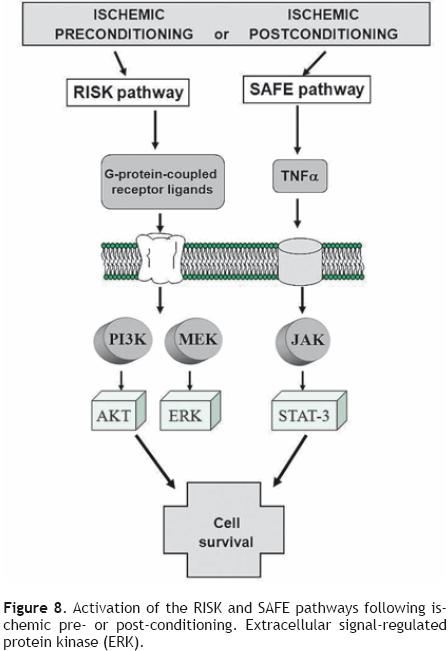
RISK pathway
During the early stages of reperfusion, there is an up–regulation of pro–survival kinases termed: the Reperfusion Injury Salvage Kinase (RISK) pathway, which, recently, has promoted a renewed interest in cardioprotective reperfusion strategies40 (Figure 3). While the mechanisms involved in reperfusion injury are known to involve apoptosis and necrosis, among others, it is also realized that cells have an inherent program for survival after ischemia–reperfusion insults, via the recruitment of innate prosurvival kinase cascades. This pathway comprises to PI3–kinases and p42/p44 extra–cellular signal–regulated kinases (ERK 1/2), which could be activated by ischemia–reperfusion injury or by PostC or pharmacological PostC. The PI3–kinases (PI3Ks) are a family of enzymes involved in several functions such as cell growth, proliferation, differentiation, motility, survival and intracellular trafficking. These enzymes are capable of phosphorylating the 3–position hydroxyl group of the inositol ring of phosphatidylinositol. Particularly, the PI3K–Akt (Akt = protein kinase B) and MEK kinases (MEK kinase = MAPK/ERK kinase; mitogen–activated protein kinase/extracellular signal–regulated kinase) have shown to be important components of the cell survival pathway and have antiapoptotic effects. There are several potential mechanisms, none of them mutually exclusive, through which this pathway mediates inhibition of mPTP opening. These are: 1) phosphorylating and inhibiting GSK3β;41 2) phosphorylating eNOS and producing nitric oxide, which has been demonstrated to inhibit mPTP opening42; and 3) phosphorylating BAD, either directly or indirectly via p70S6 kinase,33,43,44 thereby negating its proapoptotic effect. Recently, Davidsson and colleagues.45 found that insulin–mediated phosphorylation of PI3K–Akt protects the myocyte against oxidative stress by inhibiting mPTP. This suggests that the pharmacological activation of the RISK pathway by insulin at the onset of reperfusion protects against ischemia–reperfusion injury by reducing the probability that the mPTP will open. This mechanism could participate in the cardioprotective effect of "GIK" solution, which is composed by insulin in combination with glucose and potassium. However, clinical studies have suggested that the GIK solution may be cardioprotective following myocardial ischemia, via a direct, non–metabolic cardioprotective effect.46, 47 Albeit, whether the RISK pathway kinases and the mPTP are involved in the clinical setting remains to be examined. At this moment, it is necessary to ascertain the potential mechanism whereby the RISK pathway delays mPTP opening and protects the cells from injury, and this issue is part of the setting of preconditioning or protection against reperfusion–induced injury in experimental models.
Postconditioning the human heart
The ultimate validation and utility of a cardioprotective therapy is its application to humans presenting with coronary artery disease and concomitant risk factors. However, at this date there are very few studies conducted in humans; this situation is understandable because of the high risks posed by this approach. Two studies have recently reported that conventional PostC is an effective treatment in a select patient population with coronary artery disease. In a study by Laskey,48 17 patients undergoing PCI were enrolled to receive standard angioplasty involving 90s of uninterrupted balloon inflation without further treatment (n = 7) or repeated balloon inflation ("conditioning", n = 10) of 90 s duration applied 3 to 5 min after the angioplasty inflation. "Conditioning" after angioplasty reduced the magnitude of ST–segment elevation compared to controls, and accelerated the rate at which ST elevation normalized after reperfusion. Furthermore, blood flow velocity reserve was significantly improved in "conditioned" hearts. Staat and colleagues 49 reported a multi–center randomized clinical trial in 37 patients with total coronary artery occlusion undergoing angioplasty/ stenting. Patients that achieved a TIMI flow grade of 2 to 3 at completion of the angioplasty/stent procedure were randomized to receive either standard treatment thereafter or PostC with 4 cycles of 1–min re–inflation, followed by 1–min deflation of the angioplasty balloon. Infarct size was significantly less, and the coronary blood flow achieved was greater in the postconditioned patients. There were no adverse events in the postconditioned patients. Together, these two studies suggest that PostC represents a safe and efficient cardioprotective intervention for the treatment of reperfusion injury in patients with ischemic heart disease. These data must be reproduced by other clinical trials, and in broader patient populations, particularly those presenting with severe coronary artery disease involving high risk factors (hypertension, hypercholesterolemia, obesity, diabetes).
Pharmacological PostC
Despite the evidence mentioned above, in the clinical situations, ischemic PostC may be conceptually difficult to introduce, i.e., reintroduction of ischemia at the time of reperfusion may lead to potential complications. These complications are the cause of the initial refusal to use this approach in clinical settings. For example, during primary angioplasty, repetitive inflations and deflations of the balloon may result in coronary plaque rupture with consequences for restenosis or embolic events. During coronary bypass surgery, interruptions to reperfusion via the newly grafted conduit may only lead to regional myocar–dial protection in the area supplied by that bypass. An alternative strategy during bypass surgery would be repetitive clamping and unclamping of the ascending aorta to achieve ischemic PostC, a concept, however, that many cardiac surgeons would be unwilling to perform due to the high risk of disrupting atheromatous plaque debris and subsequent risk of stroke. The use of ischemic PostC would not be possible in an acute myocardial infarction patient referred for thrombolysis, in case of unstable angina, in patients presenting with non–ST segment elevation myocardial infarction or in the event of a cardiac arrest. Such a protocol might be possible to apply in a patient with myocardial infarction referred for PCI, elective PCI, or at the time of cardiac surgery. However, the concept of "pharmacological PostC" by the administration of agents that activate the RISK pathway and mediate the protective effects of PostC is a more practical solution. Agents such as insulin,44 atorvastatin,50 bradikinin,51 tumor growth factor (TGF)–B,52 and glucagon–like peptide 1 (GLP–1),53 anesthetics,54–59 opiods60–63 could individually be used as adjuvant to current reperfusion strategies, such as thrombolytics and primary PCI, to limit lethal reperfusion injury and could form the basis of much needed and important reperfusion strategies.
Anesthetic postconditioning (APostC)
Experimental evidence indicates that volatile anesthetics are capable of exerting cardioprotective effects under these conditions. For example, halothane prevented reoxygenation–induced hypercontracture of cardiac myocytes in vitro, a potential cause of myocyte necrosis during early reperfusion.64 Halothane also reduced reperfusion injury after regional myocardial ischemia in rabbit hearts.65 Desflurane and sevoflurane reduced infarct size when administered during the first 15 minutes of reperfusion in rabbits.66 Isoflurane enhanced the functional recovery of isolated rat hearts when administered solely during reperfusion.67 The administration of sevoflurane after ischemia also improved contractile and metabolic function concomitant with reduced myoplastic calcium loading in isolated guinea pig hearts.68 These and other findings indicate that volatile anesthetics may prevent intracellular calcium overloading during early reperfusion, presumably by virtue of their actions as voltage–dependent calcium channel antagonist. Isoflurane and sevoflurane also reduced postischemic adhesion of neutrophils,69 an important source of oxygen–derived free radicals during reperfusion that are known to be critical mediators of reperfusion injury.70 Several protective effects of anesthetics inhalation have been proposed (Figure 4).
Opioid and postC
Activation of G protein–coupled receptors such as adenosine and opioid receptors has been demonstrated to initiate preconditioning. In the case of PostC, the G protein–coupled receptor activation may serve as essential mechanism that also triggers protection. Indeed, recent reports have addressed that adenosine A2a and A2b receptors are responsible for the protection granted by PostC.71 Recently, Gross and colleagues, demonstrated that opioids can reduce infarct size when administered just before reperfusion, an effect that was similar to that observed when opioids were given before ischemia.72 Furthermore, Weihrauch and colleagues73 reported that morphine enhanced the isoflurane–induced PostC in rabbit hearts. Therefore, it is possible that PostC protects the heart by activating opioid receptors during reperfusion. On the other hand, activation of delta opioid receptors plays an important role in PostC and may protect the heart at reperfusion by modulating the mPTP opening via a signaling cascade involving delta and kappa opioid receptors, NO, PKC, and other pathways (Figure 5). Gross and colleagues72 and Fryer and colleagues,74 showed that opioid–induced PostC was mediated by activation of G protein–coupled delta 1 opioid receptors, PI3K–mediated signaling, and mitoKA–TP channels. Two selective delta 1 opioid receptor agonists (TAN–67 and BW373U86) and morphine were initially shown to augment isoflurane preconditioning in rats,75 and the nonselective opioid antagonist, naloxone, inhibited these beneficial actions. Interestingly, naloxone pretreatment also abolished reductions in infarct size produced by administration of isoflurane alone before ischemia and reperfusion, showing for the first time that opioid receptors mediate anesthetic preconditioning. Our group, at the pharmacology department at Instituto Nacional de Cardiología Ignacio Chávez, showed that remifentanil, an agonist of opioid receptors, elicited antiarrhythmic and cardioprotective effects in experimental ventricular arrhythmias induced by digoxin and, in a model of two atrial arrhythmias induced by aconitine and by electrical stimulation.76
Glycine cardioprotection
The amino acid, glycine (Gly), has been shown to exert a protective effect against cell death after diverse insults including ischemia–reperfusion injury in different cell types other than cardiomyocytes77 (Figure 6). Gly is an inhibitory neurotransmitter in the central nervous system that has been demonstrated by a wide range of studies in the past few years. Gly prevents or mitigates a variety of pathological processes in experimental models, such as inflammation, shock, endotoxemic shock, and those associated with transplantation. The classic inhibitory gly receptor (GlyR) in the central nervous system is a ligandgated membrane–spanning ion channel.78,79 GlyR consists of three types of subunits: (alpha), a ligand binding subunit; (beta), a structural subunit; and gephyrin, a cytoplasmic anchoring protein.80 Gly in the central nervous system exerts its inhibitory actions by binding the GlyR that is located mainly on the postsynaptic neuronal membranes. Activation of GlyR leads to increase in chloride conductance.81 An influx of chloride hyperpolarizes postsynaptic membranes, thus counteracting the depolarizing action of excitatory neurotransmitters. It is proposed that hyperpolarization decreases the opening of calcium channels, thereby blocking the movement of calcium across the plasma membrane and the subsequent events in the inflammatory process. Recent studies have provided pharmacological and molecular evidence for the existence of GlyR in endothelial cells,82 renal proximal tubular cells,83 and most leukocytes.84 Several investigators reported the benefits of N–(2–mercaptopropionyl)–glycine (MPG), a hydroxyl radical scavenger, for ischemic–reperfused hearts; treatment of anesthetized animals with this agent reduced ischemia–reperfusion–induced formation of the infarct size,85 and improved ischemia–reperfusion induced myocardial stunning.86 Furthermore, treatment of perfused hearts with MPG enhanced recovery of the contractile function and preserved the ultrastructural integrity of the myocardium.87 Ruiz–Meana's and colleagues showed that the amino acid Gly has a powerful and previously unrecognized inhibitory effect on mPTP, similar of that of low pH, in rat heart mitochondria. They also demonstrated for the first time that addition of Gly during reoxygenation prevents acute necrotic cell death associated with pH normalization in cultured cardiac myocytes and LDH release in isolated rat hearts. Moreover, exposure to Gly during reoxygenation not only reverses cellular Gly depletion observed in cells subjected to ischemia–reoxygenation, but markedly raises intracellular Gly content up to the range proven to inhibit mPTP in isolated mitochondria.88
Postconditioning the human heart with adenosine
Adenosine is a primary mediator of postconditioning effects. Studies performed in animal models indicate that adenosine receptor activation is involved in the cardioprotection conferred by PostC. The underlying mechanism may involve delaying the washout of endo–genously released adenosine during the early minutes of reperfusion.89 The protective effects could be blocked by the nonselective adenosine receptor antagonist, 8SPT,90 and by A2a and A3 selective antagonists given before PostC. Adenosine has also been implicated in the cardioprotection exerted by remote postC.91 In a study of 60 patients with rheumatic heart valve disease undergoing heart valve replacement operations, patients were randomized to an adenosine (1.5 mg/kg) or saline (control) bolus injection through an arterial catheter immediately after the aorta cross–clamp had been removed. The results of this study were the inotrope scores in the intensive care unit (ICU) were much lower, and the ICU time was significantly shorter in the adenosine group. More important, cardiac troponin I release was lower, especially at 12 and 24 hours after reperfusion. That study demonstrated that pharmacologic PostC with adenosine produces protective effects against myocardial reperfusion injury in cardiac operations.92
Advances in PostC
Activation of the protective survivor activating factor enhancement (SAFE) pathway against reperfusion injury. Novel strategies to protect the heart during myocardial reperfusion have emerged as very promising experimental therapies against lethal reperfusion injury. One of them, is the activation of the protective survivor activating factor enhancement (SAFE) pathway.93 This path includes the participation of TNFα and STAT–3. TNFα levels (blood and tissue) are increased in acute myocardial infarction and, generally, TNFα is implicated as a mediator of adverse remodeling in heart failure.94 TNFα exerts its action after binding onto its cell surface receptors, TNFα receptor 1 (TNFR1 or p55) and TNFα receptor 2 (TNFR2 o p75). Both receptor subtypes are found in human and rat cardiac myocytes.95 There are controversial findings that have been described with beneficial and detrimental effects for TNFα.96,97 These differences about its effects can be explained depending on which TNFα receptor is activated. Thus, TNFα 1 may be cardiotoxic whereas TNFα receptor 2 could be cardioprotective. Lecour and colleagues,98 have demonstrated that exogenous TNFα, in a dose and time dependent manner, could mimic ischemic preconditioning in the rat or mouse model.
The mechanisms proposed for the protective effect of TNFα is the following: After activation of the membrane receptors by TNFα (other receptors could be participating, like interleukin 6 or growth factors receptors), two adjacent juxtaposed JAKs (Janus kinase) are transphosphorylated and subsequently activate the STAT pathway. JAKs are a family of tyrosine kinases that are associated with membranes receptors and play a major role in the transduction of signals from the cytosol to the nucleus, and they are activated during ischemia and ischemia reperfusion in the heart. The STAT family of transcription factor proteins consists of seven identified members: STAT–1, STAT–2, STAT–3, STAT–4, STAT–5A, STAT–5B, and STAT6. After activation with JAKs, STATs form homo– or heterodimers that are translocated to the nucleus, resulting in gene transcription (Figure 7). The activation of the JAK/ STAT pathway plays a crucial role in the expression of stress–responsive genes that could be participating in the protective effects on the heart.99
Acting together SAFE and RISK pathways
These distinct pathways are activated at the time of reperfusion following an ischemic preconditioning insult, but whether these two pathways may interact together or be totally independent remains unclear. Lecour's group has found that TNFα–mediated preconditioning acts independently of the activation of the RISK pathway, therefore suggesting that activation of both pathways at the time of reperfusion is not always mandatory to protect the heart against reperfusion injury.98 However, the inhibition of either the RISK or the SAFE pathway in ischemic preconditioning totally abolished the protection in ischemic preconditioning, suggesting the possibility of a cross–talk between the two pathways at the level of PI3K/Akt and JAK/STAT–3.98–100 (Figure 8).
The precise molecular mechanisms and pathways of signaling protective cascades are far from being fully elucidated. However, there is now strong evidence for the existence of at least two pathways with TNFα that play a key role in the immune system. Therefore, they represent a novel and alternative protective SAFE path during both ischemic pre– and post–conditioning.
It is clear now that it is necessary to gather more experimental evidence, besides those mentioned above, in order to encourage the development of minimal risk procedures that could be used in the clinical setting.
Conclusion
Although many of the pathways involved in PostC have also been identified in IPC, recent evidences shows that both processes could be different in their pathways, being the end–effector the mPTP opening. Thus, we could get benefits from the different timing of these approaches and apply the PostC at the onset of reperfusion in a clinical setting (angioplasty, cardiac surgery, transplantation, etc). The PostC represents a new therapeutic tool that could be considered part of existing therapeutic resources. The experimental evidence reviewed in this work supports its possible clinical application in an immediate future, albeit, more randomized, double–blind, and large–scale studies are needed. We have no doubts that in coming years this therapeutic objective will become widely accepted.
References
1. Lloyd–Jones D, Adams RJ, Brown TM, et al. Heart disease and stroke statistics–2010 update: a report from the American Heart Association Circulation 2010;121:e46–e215. [ Links ]
2. Gibbons RJ, Valeti US, Araoz PA, et al. The quantification of the infarct size. J Am Coll Cardiol. 2004;44:1533–1542. [ Links ]
3. McGovern PG, Pankow JS, Shahar E, et al. Recent trends in acute coronary heart disease–mortality, morbidity, medical care and risk factors. The Minnesota Heart Survey Investigators. N Engl J Med 1996;334:884–890. [ Links ]
4. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med 2007;357:1121–1135. [ Links ]
5. Jennings RB, Sommers HM, Smyth GA, et al. Myocardial necrosis induced by temporary occlusion of a coronary artery in the dog. Arch Pathol 1960;70:68–78. [ Links ]
6. Wu KC, Zerhouni EA, Judd RM, et al. Prognostic significance of microvascular obstruction by magnetic resonance imaging in patients with acute myocardial infarction. Circulation 1998;97:765–772. [ Links ]
7. Das AM, Harris DA. Regulation of the mitochondrial ATP synthase in intact rat cardiomyocytes. Biochem J 1990;266:355–361. [ Links ]
8. Dennis S C, Gevers W, Opie LH. Protons in ischemia: where do they come from; where do they go? J Mol Cell Cardiol 1991;23:1077–1086. [ Links ]
9. Schlüter KD, Jakob G, Ruiz–Meana M, et al. Protection of reoxygenated cardiomyocytes against osmotic fragility by NO donors. Am J Physiol 1996;271:H428–H434. [ Links ]
10. Piper HM, Abdallah Y, Schafer C. The first minutes of reperfusion: a window of opportunity for cardioprotection. Cardiovasc Res 2004; 61:365–371. [ Links ]
11. Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J 1999;341:233–249. [ Links ]
12. Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion –a target for cardioprotection. Cardiovasc Res 2004;61:372–385. [ Links ]
13. Di Lisa F, Menabo R, Canton M, et al. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem 2001;276:2571–2575. [ Links ]
14. Griffiths EJ, Halestrap AP. Mitochondrial non–specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J 1995;307:93–98. [ Links ]
15. Cohen MV, Yang XM, Downey JM. Acidosis, oxygen, and interference with mitochondrial permeability transition pore formation in the early minutes of reperfusion are critical to postconditioning's success. Basic Res Cardiol 2008;103;464–471. [ Links ]
16. Jordan JE, Zhao ZQ, Vinten–Johansen J. The role of neutrophils in myocardial ischemia–reperfusion injury. Cardiovasc Res 1999; 43: 860–878. [ Links ]
17. Engler RL, Schmid–Schönbein GW, Pavelec RS. Leukocyte capillary plugging in myocardial ischemia and reperfusion in the dog. Am J Pathol 1983;111:98–111. [ Links ]
18. Moore KL, Patel KD, Bruel RE. P–selectin glycoprotein lig–and–1 mediates rolling of human neutrophils on P–selectin. J Cell Biol 1995;128:661–671. [ Links ]
19. Zhao ZQ, Corvera JS, Halkos ME. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol 2003;285:H579–H588. [ Links ]
20. Kin H, Zatta AJ, Lofye MT, et al. Postconditioning reduces infarct size via adenosine receptor activation by endogenous adenosine. Cardiovasc Res 2005;67:124–133. [ Links ]
21. Halkos ME, Kerendi F, Corvera JS, et al. Myocardial protection with postconditioning is not enhanced by ischemic preconditioning. Ann Thorac Surg 2004;78:961–969. [ Links ]
22. Sun HY, Wang NP, Halkos M. Postconditioning attenuates cardiomyocyte apoptosis via inhibition of JNK and p38 mitogen activated protein kinase signaling pathways. Apoptosis 2006;11:1583–1593. [ Links ]
23. Yang X–M, Proctor JB, Cui L, et al. Multiple, brief coronary occlusions during early reperfusion protect rabbit hearts by targeting cell signaling pathways. J Am Coll Cardiol 2004;44:1103–1110. [ Links ]
24. Yang X–M, Philipp S, Downey JM, et al. Postconditioning's protection is not dependent on circulating blood factors or cells but involves adenosine receptors and requires PI3Kinase and guanylyl cyclase activation. Basic Res Cardiol 2005;100:57–63. [ Links ]
25. Tsang A, Hausentoy DJ, Mocanu MM, et al. Postconditioning a form of "modified reperfusion" protects the myocardium by activating the phosphatidylinositol 3–kinase–akt pathway. Circ Res 2004;95:230–232. [ Links ]
26. Zatta AJ, Kin H, Lee G, et al. Infarct–sparing effect of myocardial postconditioning is dependent on protein kinase C signaling. Cardiovasc Res 2006;70:315–324. [ Links ]
27. Jang Y, Xi J, Wang H, et al. Postconditioning prevents reperfusion injury by activating 5–opioid receptors. Anaesthesiology 2006;108:243–250. [ Links ]
28. Griffiths EJ, Halestrap AP. Protection by cyclosporin A of ischemia/reperfusion–induced damage in isolated rat hearts. J Mol Cell Cardiol 1993;25:1461–1469. [ Links ]
29. Argaud I, Gateau–Roesch O, Risky O, et al. Postconditioning inhibits mitocondrial permeability transition. Circulation 2005;111:194–197. [ Links ]
30. Philipp S, Downey JM, Cohen MV. Postconditioning must be initiated in less than 1 minute following reperfusion and is dependent on adenosine receptors and PI3–kinase. Circulation 2004;110(Suppl 111): 804–806. [ Links ]
31. Hausenloy DJ, Duchen MR, Yellon DM. Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia–reperfusion injury. Cardiovasc Res 2003; 60: 617–625. [ Links ]
32. Hausenloy DJ, Maddock HL, Baxter GF, et al. Inhibiting mitochondrial permeability transition pore opening: a new paradigm for myocardial preconditioning? Cardiovasc Res 2002;55:534– 543. [ Links ]
33. Jonassen AK, Sack MN, Mjos OD, et al. Myocardial protection by insulin at reperfusion requires early administration and is mediated via Akt and p70s6 kinase cell–survival signaling. Circ Res 2001;89:1191–1198. [ Links ]
34. Sun K, Liu ZS, Sun Q. Role of mitochondria in cell apoptosis during hepatic ischemia–reperfusion injury and protective effect of ischemic postconditioning. World J Gastroenterol 2004; 10:1934–1938. [ Links ]
35. Argaud L, Gateau–Roesch O, Raisky O, Loufouat J, Robert D and Ovize M. Postconditioning inhibits mitochondrial permeability transition. Circulation 2005; 111: 194–197. [ Links ]
36. Argaud L, Gomez L, Gateau–Roesch O, et al. Trimetazidine inhibits mitochondrial permeability transition pore opening and prevents lethal ischemia–reperfusion injury. J. Mol Cell Cardiol 2005;39:893–899. [ Links ]
37. Baines CP, Kaise RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005;434;658–662. [ Links ]
38. Nakagawa T, Shimizu S, Watanabe T, et al. Cyclophilin D–dependent mitochondrial permeability transition regulates some necrotic but no apoptotic cell death. Nature 2005;434:652–658. [ Links ]
39. Lim SY, Davidson SM, Hausenly DJ et al. Preconditioning and postconditioning: the essential role of the mitochondrial permeability transition pore. Cardiovasc Res 2007;75:530–535. [ Links ]
40. Hausenby DJ, Yellon DM. New directions for protecting the heart against ischaemia–reperfusion injury: targeting the reperfusion injury salvage kinase (RISK)–pathway. Cardiovasc Res 2004; 61: 448–460. [ Links ]
41. Juhaszova M, Zorov DB, Kim SH, et al. Glycogen synthase kinase–3 beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest 2004;113:1535–1549. [ Links ]
42. Kim JS, Ohshima S, Pediaditakis P, et al. Nitric oxide protects rat hepatocytes against reperfusion injury mediated by the mitochondrial permeability transition. Hepatology 2004;39:1533–1543. [ Links ]
43. Jonassen AK, Mjos OD, Sack MN. p70s6 kinase is a functional target of insulin activated Akt cell–survival signaling. Biochem Biophys Res Commun 2004; 315:160–165. [ Links ]
44. Jonassen AK, Brar BK, Mjos OD, et al. Insulin administered at reoxygenation exerts a cardioprotective effect in myocytes by a possible anti–apoptotic mechanism. J Mol Cell Cardiol 2000;32:757–764. [ Links ]
45. Davidson SM, Hausenloy D, Duchen MR, et al. Signalling via the reperfusion injury signalling kinase (RISK) pathway links closure of the mitochondrial permeability transition pore to cardioprotection. The International Journal of Biochemistry & Cell Biology 2006;38:414–419. [ Links ]
46. Doenst T, Bothe W, Beyersdorf F. Therapy with insulin in cardiac surgery: Controversies and possible solutions. Ann Thorac Surg 2003;75:S721–728. [ Links ]
47. Sack MMN Yellon DM. Insulin therapy as an adjunct to reperfusion after acute coronary ischemia: A proposed direct myocardial cell survival effect independent of metabolic modulation. J Am Coll Cardiol 2003;41:1404–1407. [ Links ]
48. Laskey WK. Brief repetitive balloon occlusions enhance reperfusion during percutaneous coronary intervention for acute myocardial infarction: a pilot study. Catheter Cardiovasc Interv 2005;65:361–367. [ Links ]
49. Staat P, Rioufol G, Piot C, et al. Postconditioning the human heart. Circulation 2005;112:2143–2148. [ Links ]
50. Bell RM, Yellon DM. Atorvastatin, administered at the onset of reperfusion and independent of lipid lowering, protects the myocardium by up–regulating a pro–survival pathway. J Am Coll Cardiol 2003;41:508–515. [ Links ]
51. Bell RM, Yellon DM. Bradykinin limits infarction when administered as an adjunct to reperfusion in mouse heart: the role of PI3k, Akt and eNOS. J Mol Cell Cardiol 2003;35:185–193. [ Links ]
52. Baxter GF, Mocanu MM, Brar BK, et al. Cardioprotective effects of transforming growth factor–beta 1 during early reoxygenation of reperfusion are mediated by p42/p44 MAPK. J Cardiovasc Pharmacol 2001;38:930–939. [ Links ]
53. Nikolaidis LA, Mankad S, Sokos GG, et al. Effects of glucagonlike peptide–1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation 2004;109:962–965. [ Links ]
54. Shlack W, Preckel B, Barthel H. Halothane reduces reperfusion injury after regional ischaemia in the rabbit heart in vivo. Br J Anaesth 1997;79:88–96. [ Links ]
55. Preckel B, Schlack W, Comfere T. Effects of enflurane, isoflurane, sevoflurane and desflurane on reperfusion injury after regional myocardial ischaemia in the rabbit heart in vivo. Br J Anaesth 1998; 81: 905–912. [ Links ]
56. Schlack W, Preckel B, Stunneck D. Effects of halothane, enflurane, isoflurane, sevoflurane and desflurane on myocardial reperfusion injury in the isolated rat heart. Br J Anaesth 1998;81:913–919. [ Links ]
57. Varadarajan SG, An J, Novalija E. Sevoflurane before or after ischemia improves contractile and metabolic function while reducing myoplasmic Ca2 loading in intact hearts. Anesthesiology 2002;96:125–133. [ Links ]
58. HeindI B, Reichle FM, Zahler S. Sevoflurane and isoflurane protect the reperfused guinea pig heart by reducing postischemic adhesion of polymorphonuclear neutrophils. Anesthesiology 1999;91:521–530. [ Links ]
59. Vinten–Johansen J. Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc Res 2004;61:481–497. [ Links ]
60. Jang Y, Xi J, Wang H, et al. Postconditioning prevents reperfusion injury by activating 5–opioid receptors. Anaesthesiology 2006;108:243–250. [ Links ]
61. Gross ER, Hsu AK, Gross GJ. Opioid–induced cardioprotection occurs via glycogen synthase kinase β inhibition during reperfusion in intact rat hearts. Circ Res 2004;94:960–966. [ Links ]
62. Weihrauch D, Riotikowski JG, Bienengracher M, et al. Morphine enhances isoflurane–induced postconditioning against myocardial infarction: The role of phosphatidylinositol–3–kinase and opioid receptors in rabbits. Anesth Analg 2005;101:942–949. [ Links ]
63. Gross ER, Hsu AK, Gross GJ. GSK3β inhibition and KATP channel opening mediate acute opioid–induced cardioprotection at reperfusion. Basic Res Cardiol 2007;102: 341–349. [ Links ]
64. Siegmund B, Schlack W, Ladilov YV. Halothane protects cardiomyocytes against reoxygenation–induced hypercontracture. Circulation 1997;96: 4372–4379. [ Links ]
65. Shlack W, Preckel B, Barthel H. Halothane reduces reperfusion injury after regional ischaemia in the rabbit heart in vivo. Br J Anaesth 1997;79: 88–96. [ Links ]
66. Preckel B, Schlack W, Comfere T. Effects of enflurane, isoflurane, sevoflurane and desflurane on reperfusion injury after regional myocardial ischaemia in the rabbit heart in vivo. Br J Anaesth 1998;81:905–912. [ Links ]
67. Schlack W, Preckel B, Stunneck D. Effects of halothane, enflurane, isoflurane, sevoflurane and desflurane on myocardial reperfusion injury in the isolated rat heart. Br J Anaesth 1998;81:913–919. [ Links ]
68. Varadarajan SG, An J, Novalija E. Sevoflurane before or after ischemia improves contractile and metabolic function while reducing myoplasmic Ca2+ loading in intact hearts. Anesthesiology 2002;96:125–133. [ Links ]
69. HeindI B, Reichle FM, Zahler S. Sevoflurane and isoflurane protect the reperfused guinea pig heart by reducing postischemic adhesion of polymorphonuclear neutrophils. Anesthesiology 1999;91:521–530. [ Links ]
70. Vinten–Johansen J. Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc Res 2004;61:481–497. [ Links ]
71. Kin H, Zatta AJ, Lofye MT, et al. Postconditioning reduces infarct size via adenosine receptor activation by endogenous adenosine. Cardiovasc Res 2005;67:124–133. [ Links ]
72. Gross ER, Hsu AK, Gross GJ. Opioid–induced cardioprotection occurs via glycogen synthase kinase β inhibition during reperfusion in intact rat hearts. Circ Res 2004;94:960–966. [ Links ]
73. Weihrauch D, Riotikowski JG, Bienengracher M, et al. Morphine enhances isoflurane–induced postconditioning against myocardial infarction: The role of phosphatidylinositol–3–kinase and opioid receptors in rabbits. Anesth Analg 2005;101:942–949. [ Links ]
74. Fryer RM, Wang Y, Hsu AK. Essential activation of PKC–delta in opioid–initiated cardioprotection. Am J Physiol Heart Circ Physiol 2001;280: H1346–H1353. [ Links ]
75. Ludwig LM, Patel HH, Gross GJ. Morphine enhances pharmacological preconditioning by isoflurane: Role of mitochondrial K (ATP) channels and opioid receptors. Anesthesiology 2003;98:705–711. [ Links ]
76. Luna–Ortiz P, Zarco–Olvera G, Ramírez–Ortega M, et al. Antiarrhythmic and cardioprotective effects of remifentanil in anesthetized dogs. Arch Cardiol Mex 2009;79:182–188. [ Links ]
77. Nishimura Y, Lemasters JJ. Glycine blocks opening of a death channel in cultured hepatic sinusoidal endothelial cells during chemical hypoxia. Cell Death Differ 2001; 8: 850–858. [ Links ]
78. Rajendra S, Lynch JW, Schofield PR. The glycine receptor. Pharmacol Ther 1997;73:121–146. [ Links ]
79. Laube B, Maksay G, Schemm R, et al. Modulation of glycine receptor function: a novel therapeutic approach for therapeutic intervention at inhibitory synapses? Trends Pharmacol Sci 2002;23:519–527. [ Links ]
80. Pfeiffer F, Betz H. Solubilization of the glycine receptor from rat spinal cord. Brain Res 1981;226:273–279. [ Links ]
81. Werman R, Davidoff RA, Aprison MH. Is glycine a neurotransmitter? Nature 1967;214:681–683. [ Links ]
82. Yamashima S, Konno A, Wheeler MD, et al. Endothelial cells contain a glycine–gated chloride channel. Nutr Cancer 2001;40:197–204. [ Links ]
83. Reeves WB. Effects of chloride channel blockers on hypoxic injury in rat proximal tubules. Kidney Int 1997;51:1529–1534. [ Links ]
84. Ikejima K, Limuro Y, Forman DT, et al. A diet containing glycine improves survival in endotoxic shock in the rat. Am J Physiol 1996;271:G–97–103. [ Links ]
85. Koerner JE, Anderson BA, Dage RC. Protection against postischemic myocardial dysfunction in anesthetized rabbits with scavengers of oxygen–derived free radicals. J Cardiovasc Pharmacol 1991;17:185–191. [ Links ]
86. Myers ML, Bolli R, Lekich RF, Hartley et al. N–2mercaptopropionylglycine improves recovery of myocardial function after reversible regional ischemia. J Am Coll Cardiol 1986;8:1161–1168. [ Links ]
87. Tanaka M, Fujiwara H, Yamasaki K, et al. Superoxide dismutase and N–2 mercaptopropionylglycine attenuate infarct size limitation effect of ischemic preconditioning in the rabbit. Cardiovasc Res 1994;28:980–986. [ Links ]
88. Ruiz–Meana M, Pina P, Garcia–Dorado D, et al. Glycine protects cardiomyocytes against lethal reoxygenation injury by inhibiting mitochondrial permeability transition. J Physiol 2004;558:873– 882. [ Links ]
89. Kin H, Zatta AJ, Lofye MT, et al. Postconditioning reduces infarct size via adenosine receptor activation by endogenous adenosine. Cardiovasc Res 2005;67:124–133. [ Links ]
90. Yang XM, Philipp S, Downey JM, et al. Postconditioning protection is not dependent on circulating blood factors. Basic Res Cardiol 2005;100:57–63. [ Links ]
91. Kerendi F, Kin H, Halkos ME, et al. Remote postconditioning: brief renal ischemia and reperfusion applied before coronary artery reperfusion reduces myocardial infarct size via endogenous activation of adenosine receptors. Bas Res Cardiol 2005;100:404–412. [ Links ]
92. Jin ZX, Zhou JJ, Xing M. Postconditioning the human heart with adenosine in heart valve replacement surgery. Ann thorac Surg 2007;83:2066–2073. [ Links ]
93. Lecour S. Multiple protective pathways against reperfusion injury: a SAFE path without Aktion? J Mol Cell Cardiol 2009;46:607– 609. [ Links ]
94. Mann DL. Stress–activated cytokines and the heart: from adaptation to maladaptation. Annu Rev Physiol 2003;65:81–101. [ Links ]
95. Torre–Amione G, Kapadia S, Lee J, et al. Expression and functional significance of tumor necrosis factor receptors in human myocardium. Circulation 1995;92:1487–1493. [ Links ]
96. Kurreimeyer KM, Michael LH, Baumgarten G, et al. Endogenous tumor necrosis factor protects the adult cardiac myocyte against ischemic–induced apoptosis in a murine model of acute myocardial infarction. Proc Natl Acad Sci USA 2000;97:5456–5461. [ Links ]
97. Flaherty MP, Guo Y, Tiwari S, et al. The role of TNF–alpha receptors p55 and p75 in acute myocardial ischemia/reperfusion injury and late preconditioning. J Mol Cell Cardio 2008;45:735–741. [ Links ]
98. Lecour S, Suleman N, Deuchar GA, et al. Pharmacological preconditioning with tumor necrosis factor–alpha activates signal transducer and activator of transcription–3 at reperfusion without involving classic prosurvival kinases (Akt and extracellular signal–regulated kinase). Circulation 2005;112:3911–3918. [ Links ]
99. Boengler K, Hilfiker–Kleiner D, Drexler H, Heusch G, Schulz R. The myocardial JAK/STAT pathway: from protection to failure. Pharmacol Ther 2008;120:172–185. [ Links ]
100. Gross ER, Hsu AK, Gross GJ. The JAK/STAT pathway is essential for opioid–induced cardioprotection: JAK2 as a mediator of STAT3, Akt, and GSK–3 beta. Am J Physiol Heart Circ Physiol 2006;291:H827–834. [ Links ]

