











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
El artículo cuarto de la Constitución Política de los Estados Unidos Mexicanos señala que toda persona tiene derecho a la salud (Diario Oficial de la Federación de México (DOF), 2017). Un rubro importante de la salud es el acceso a los medicamentos, los cuales son el resultado de un proceso de innovación y desarrollo que puede alcanzar costos elevados y por tanto inaccesibles a la población. México incorporó las tendencias internacionales en materia de abasto de medicamentos desde 1977 (DOF, 1977), al publicar el Cuadro Básico de Medicamentos, un recurso importante para la administración de los insumos en hospitales del sector público.
A partir de la década de los noventa el Estado mexicano intentó abatir el alto costo de los insumos para la salud mediante la permisión de medicamentos que fueran intercambiables con los “innovadores” o “de patente”. Estos últimos son a los que se les ha otorgado una patente que le confiere exclusividad de producción y procesos industriales por un tiempo de 20 años para el caso de México (Instituto Mexicano de la Propiedad Intelectual (IMPI), 2016).
Sin embargo, el lanzamiento de estos productos se llevó a cabo en un marco regulatorio deficiente; esto confluyó con el sistema económico capitalista imperante en el país. Ambas situaciones abrieron paso a una cultura mal informada acerca de los medicamentos genéricos en México. A continuación damos un ejemplo de esta confusión.
El 8 de septiembre de 1997 se constituyeron las Farmacias Similares® “con la finalidad de ofrecer productos y servicios de salud a los estratos más desprotegidos del país” (Farmacias Similares, 2017). Los años siguientes se lanzó una potente campaña mediática que publicitaba los beneficios de los medicamentos “similares”. La población, que no conocía la diferencia entre medicamentos innovadores y genéricos, fue introducida a los “medicamentos similares” antes de recibir información de las autoridades sanitarias relativa a estos productos. En esa época no existía en la regulación sanitaria el concepto de los medicamentos “genéricos intercambiables”, ni una norma oficial mexicana al respecto (DOF, 1999).
La NOM-177-SSA1-1998 (DOF, 1999), Que establece las pruebas y procedimientos para demostrar que un medicamento es intercambiable. Requisitos a que deben sujetarse los terceros autorizados que realicen las pruebas instituyó el término de “medicamento genérico”, más no el de “medicamento similar”. Sin embargo, el vocablo “similar” (sólo es una acotación de la marca registrada Farmacias Similares) lo que siguió siendo usado con fines comerciales en los medios masivos de comunicación, y con el paso de los años se afianzó en el imaginario colectivo mexicano, tomándose como un sinónimo de “genérico”. Este hecho trascendió en el tiempo y forma que en el reporte sobre la industria farmacéutica (IF) publicado por la Secretaría de Economía (SE) en (ProMéxico, 2013) se menciona la existencia tanto de medicamentos genéricos como “similares”, a pesar de la ausencia en la legislación de los segundos; no obstante la SE realiza la aclaración que los medicamentos similares no cuentan con pruebas de bioequivalencia y biodisponibilidad por lo que no se garantiza su calidad.
Por otro lado, la Organización Mundial de la Salud (OMS) ha generado una gran expectativa en los medicamentos genéricos con el objetivo de promover ahorros a los Estados en el gasto que el acceso a la salud les representa. Según la Dra. Margaret Chan, directora general de esta organización: “Los países podrían ahorrarse alrededor de un 60% de gastos farmacéuticos si en lugar de medicamentos originales comprasen productos genéricos” (Chan, 2011). La OMS ha guiado sus decisiones según bases científicas, derivadas de la biofarmacia.
La producción de medicamentos genéricos es un eje prioritario en las políticas de salud pública de México, porque favorece el derecho a la salud de la población y a la vez posibilita el ahorro del gobierno en uno de los rubros de salud que más costo tienen: los medicamentos. En el DOF, 2013, el Programa Sectorial de Salud 2013-2018, indica que: “El tema de abasto de medicamentos en las instituciones del sector público está en lo más alto de la agenda gubernamental” (DOF, 2013) de hecho este Programa plantea como un objetivo: “Fomentar el ingreso de nuevos medicamentos genéricos y biocomparables seguros, de calidad y eficaces”.
La legislación mexicana favorece la inclusión de nuevos medicamentos genéricos al mercado nacional. En DOF (2016), dice que el artículo 167, fracción V del Reglamento de Insumos para la Salud establece que para obtener el registro sanitario de un medicamento genérico, el informe sobre eficacia terapéutica y seguridad del fármaco puede ser sustituido por los resultados de las pruebas de intercambiabilidad establecidos (DOF, 2013) y en el Acuerdo que determina el tipo de prueba para demostrar intercambiabilidad de medicamentos genéricos y demás disposiciones aplicables, publicado en DOF el 13 de julio de 2015.
La presente revisión muestra el desarrollo histórico del marco regulatorio en materia de medicamentos genéricos en México, resaltar el impacto que la IF tiene en la economía nacional y atraer la atención sobre la importancia de las pruebas esenciales para determinar si un medicamento es genérico.
Desarrollo económico de la industria farmacéutica en México
El envejecimiento poblacional y el aumento del acceso a los servicios de salud de la clase media han favorecido el crecimiento de la IF a nivel global. Sin embargo, las medidas de austeridad de los Estados, la expiración de las patentes de productos líder y la demanda de medicamentos genéricos son algunos factores que han afectado considerablemente las utilidades de las empresas farmacéuticas de origen europeo, australiano y japonés. Se espera que en los próximos años el consumo y producción de la IF se centren en los mercados emergentes productores de medicamentos genéricos como Rusia, China, India, Sudáfrica, México y Brasil (ProMéxico, 2013).
Desde principios de la segunda década del siglo XXI México se ha posicionado como el primer país exportador de medicamentos en América Latina (AL) (ProMéxico, 2013). Es un importante productor de antibióticos, antiinflamatorios y oncológicos. 14 de las 15 principales compañías farmacéuticas a nivel internacional están situadas en el país (ProMéxico, 2013), (en el año 2008 se eliminó de la Ley General de Salud (LGS) el requisito de planta para obtener un registro sanitario). El sector representa el 0.48 % del Producto Interno Bruto (PIB) y el 2.9 % del PIB manufacturero (ProMéxico, 2015). Compárese, por ejemplo, con la industria refresquera, que representó en el mismo año el 0.5% del PIB y el 2.8% del PIB manufacturero (Cable News NetworicExpansión (CNNExpansión), 2015).
En el 2013 el sector farmacéutico registró 86,783 empleos totales (Cámara Nacional de la Industria Farmacéutica (CANIFARMA), 2016). En este rubro destaca el aumento de 74% en la contratación de trabajadores con nivel de posgrado (maestría y doctorado) entre el 2007 y el 2013 (CANIFARMA, 2016); ello indica un incremento en las actividades de investigación y desarrollo (I&D) y/o en las funciones que requieren conocimiento especializado. En el mismo periodo México atrajo una inversión extranjera de más de 200 mil millones de pesos (mdp) y se verificó un crecimiento en las exportaciones a una tasa anual del 7.3%. Aunque las importaciones también crecieron, lo hicieron a una tasa anual del 4.1% (CANIFARMA, 2016), que es significativamente menor.
Según la SE en el año 2014 la producción de la IF nacional fue de 11,430 millones de dólares (mdd) y se espera que para el año 2020 alcance 21,475 mdd (ProMéxico, 2013) con una tasa media de crecimiento anual (TMCA) del 6.7% (ProMéxico, 2015). Durante el 2014 se exportó la cifra de 1,874 mdd, convirtiendo a México en el principal exportador de AL (ProMéxico, 2015). A pesar de los avances en el sector, la balanza comercial es un punto que atender, pues en este año de 2014 el país importó 4,939 mdd (ProMéxico, 2015), marcando una diferencia de 3,065 mdd.
Entre el 2005 y el 2014 países como Estados Unidos, Luxemburgo y Países Bajos hicieron una inversión de 3,152 mdd en el sector farmacéutico nacional (ProMéxico, 2015).
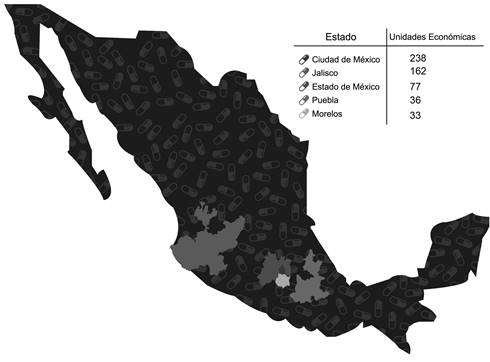
Según el Directorio Estadístico Nacional de Unidades Económicas (DENUDE), en México durante el año 2016 (Instituto Nacional de Estadística y Geografía (INEGI), 2016) se registraron 770 unidades económicas (UE) (referente a plantas de producción tanto farmacéuticas como farmoquímicas), siendo los estados con mayor presencia de la IF en el país la Ciudad de México con 238, Jalisco con 162, Estado de México con 77, Puebla con 36 y Morelos con 33 (INEGI, 2016). (Figura 1) Adicionalmente se puede observar que la IF nacional se encuentra en gran medida centralizada geográficamente.
Evaluación de medicamentos genéricos y Sistema de Clasificación Biofarmacéutica
Para realizar una evaluación comparativa de los medicamentos genéricos frente a un producto innovador o de referencia se tiene que recurrir a un conjunto de técnicas y datos que se engloban en la rama del conocimiento de la biofarmacia. Según Jung, la biofarmacia es “La disciplina que aborda la relación existente entre las propiedades fisicoquímicas del fármaco, la forma farmacéutica y la vía de administración en la velocidad y cantidad de fármaco absorbido” (Jung, 2013). En primera instancia se debe comprender que la Farmacopea de los Estados Unidos Mexicanos (FEUM) define a una forma farmacéutica como la disposición física que se le da a los fármacos y aditivos para constituir un medicamento y facilitar su dosificación y administración (Comisión Permanente de la Farmacopea de los Estados Unidos Mexicanos (FEUM), 2014a).
En complemento a la biofarmacia se encuentra la farmacocinética, que evalúa la cinética de los procesos de transferencia de los fármacos en el organismo y son: liberación, absorción, distribución, metabolismo y eliminación de los fármacos, conocido en su conjunto como proceso LADME. (Shargel, Wu-Pong & Yu, 2005). Con el fin de estudiar el proceso LADME, se recurre a algunos parámetros farmacocinéticos como el área bajo la curva (ABC) que indica la cantidad de fármaco absorbido que hay en la sangre del paciente, mientras que la concentración plasmática máxima (Cpmáx) establece el punto máximo de la absorción del fármaco así como la velocidad con la que éste alcanza la circulación sistémica (Shargel, Wu-Pong & Yu, 2005). El tiempo de vida media (t1/2) evalúa el tiempo necesario para que la mitad del fármaco sea eliminado del cuerpo del paciente, si bien no es un parámetro de bioequivalencia (Universidad Virtual de la Salud de Cuba, 2011) (Figura 2).

Figura 2: Gráfica que muestra los principales parámetros farmacocinéticos. Basada en: Lorenzo Fernández, et al. 2009; Beitia, 2009.
Una de las pruebas biofarmacéuticas más utilizadas para evaluar el desempeño de un candidato a medicamento genérico frente al innovador o de referencia es la “prueba de disolución” que determina la cantidad de fármaco que se disuelve (liberación) en un medio específico a partir de una forma farmacéutica a un tiempo determinado. Cuando se toman muestras a diferentes tiempos, bajo condiciones controladas y validadas (Baena & Ponce D’León, 2008), se le denomina prueba de perfil de disolución in vitro. La prueba de disolución es cotidianamente utilizada como una herramienta de control de calidad lote a lote mientras que el perfil de disolución se emplea para comparar diferentes lotes o diferentes productos. Incluso en algunos casos existe una correlación in vivo-in vitro (CIVIV) entre el perfil de disolución y la biodisponibilidad (Fotaki, 2011). La FEUM (Comisión Permanente de la FEUM, 2014a) propone diversos aparatos para evaluar tabletas y cápsulas mediante la prueba de disolución, en los que el volumen de medición es constante y entre los que destacan el aparato 1 (de canastillas) y el 2 (de paletas).
En 1991, la compañía Sotax Corporation (Sotax Corporation, 2016) introdujo el Aparato USP 4 (por las siglas en inglés de United States Pharmacopeia) conocido también como celda de flujo continuo (The United Satates Pharmacopeia Convention, Inc., 2004), obteniendo la patente en los Estados Unidos en 1992 (Benz, 1992). Entre sus principales características se encuentra que las condiciones sink (exceso de medio para evitar la saturación del fármaco en el mismo) (Argos Pharma, 2017) se mantienen debido a que el flujo del medio es continuo (en la modalidad de sistema abierto). Este aparato se encuentra descrito en el método general de análisis (MGA) 0521 para liberación controlada de la edición vigente de la FEUM (Comisión Permanente de la FEUM, 2014a). El mismo documento describe los aparatos 3 (cilindro oscilante), 5 (paleta sobre disco), 6 (cilindro rotatorio) y 7 (portamuestra oscilante) (Comisión Permanente de la FEUM, 2014a).
Aunque las pruebas de disolución in vitro son muy utilizadas, en algunos casos no revelan fielmente lo que sucede en el cuerpo humano, especialmente en los casos en que la absorción del fármaco está comprometida debido a su baja permeabilidad.
El sistema de clasificación biofarmacéutica (SCB) se introdujo en 1995 por Gordon Amidon (González Casado, 2015) de la Facultad de Farmacia de la Universidad de Michigan (College of Pharmacy, 2017). El SCB es un conjunto de criterios basados precisamente tanto en la permeabilidad intestinal como en la solubilidad acuosa de un fármaco, dos factores que condicionan la absorción.
Dado que el modelo permite hacer predicciones acerca de la cinética de las formas orales de liberación inmediata (Basanta, Kumar & Karunakar, 2011), fue el sustento para la Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System Guidance for Industry editada por la Food and Drug Administration (FDA) en agosto de 2000 (FDA, 2015) y en el Anexo 7 del Informe 40 del Comité de expertos de la Organización Mundial de la Salud (OMS) en el rubro de especificaciones para preparaciones farmacéuticas del 2006 (OMS, 2006).
La OMS define un fármaco “altamente soluble” como aquél que en su mayor dosis es soluble en 250 mL de medio acuoso o menos, en un intervalo de pH de 1.2 a 6.8, y a una temperatura de 37±1°C (OMS, 2006). Mientras que la FDA acepta una definición igual salvo que establece el intervalo de pH entre 1 y 6.8 (FDA, 2015).
El mismo texto de la OMS (OMS, 2006) considera un fármaco como “altamente permeable” si la cantidad absorbida determinada por un balance de masas o bien comparada con la dosis intra-venosa es mayor al 85% en humanos. El mismo criterio es usado por la FDA (FDA, 2015).
El SCB es considerado durante la investigación, desarrollo y control de calidad de los medicamentos genéricos y tiene especial relevancia porque sirve para establecer los casos en que es posible determinar la exención de pruebas in vivo a aquéllos genéricos con formas posológicas orales sólidas de liberación inmediata de nueva creación (en cuyo caso las pruebas son sustituidas por estudios de disolución in vitro con varios medios relevantes). Así, en algunos casos el SCB es usado como una herramienta para sustituir la bioequivalencia por estudios de disolución (Basanta, Kumar & Karunakar, 2011). Otra opción para poder omitir el estudio in vivo, se apoya en una hipótesis matemática que describe cuantitativamente los procesos dinámicos que ocurren entre el fármaco y el cuerpo humano. La correlación in vivo - in vitro (CIVIV) (Basanta, Kumar & Karunakar, 2011) es precisamente el modelo predictivo que describe la relación entre una característica in vitro de la forma de dosificación y una variable in vivo. Sólo se puede obtener una CIVIV cuando el proceso de absorción está limitado por la velocidad de disolución. Los parámetros más comúnmente utilizados para llevar a cabo la CIVIV son la fracción disuelta (Fd) a un tiempo para los datos in vitro y la fracción absorbida (Fa) a un tiempo para los datos in vivo (González Casado, 2015). La Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System Guidance for Industry editada por la FDA, no considera la CIVIV a partir del SCB (FDA, 2015). Algunos autores establecen que:
“Este cambio de enfoque (SCB) exige considerar la solubilidad y la permeabilidad como los factores determinantes de este proceso de absorción para así poder extrapolar, en algunos casos, ensayos realizados in vitro a comportamientos in vivo, los cuales se consolidan como una herramienta de predicción muy útil que permite disminuir la experimentación en seres humanos” (Baena & Ponce D´León, 2008).
A continuación se describen las categorías del SCB.
Categorías de fármacos según el SCB
Clase I: Alta solubilidad y permeabilidad; el paso limitante de la velocidad de absorción es la velocidad de disolución o el vaciamiento gástrico. Dentro de este grupo se encuentran los siguientes ejemplos: metoprolol, diltiazem y verapamil (Basanta, Kumar & Karunakar, 2011).
Clase II: Baja solubilidad y alta permeabilidad; presentan una absorción más lenta que los de Clase I; la biodisponibilidad de estos fármacos está limitada por su tasa de solvatación. Dentro de este grupo se encuentran por ejemplo: ketoconazol, naproxeno y nifedipino (Basanta, Kumar & Karunakar, 2011).
Clase III: Baja permeabilidad y alta solubilidad; la permeabilidad es el paso limitante. En este grupo se incluyen, entre otros: ranitidina, aciclovir y captopril (Basanta, Kumar & Karunakar, 2011).
Clase IV: Baja solubilidad y baja permeabilidad; presentan una pobre biodisponibilidad por vía oral y baja absorción en la mucosa intestinal (aunque existen algunas excepciones en esta clase de fármacos que son de administración oral). En este grupo se encuentran por citar algunos: furosemida, hidroclorotiazida y taxol (Basanta, Kumar & Karunakar, 2011).
La Figura 3 muestra las clases de fármacos según su permeabilidad y solubilidad.

El SCB es un referente en cuanto a lo que la exención de estudios in vivo se refiere, puesto que a través de la determinación de los parámetros antes descritos evita la experimentación innecesaria en humanos y ahorra a las empresas el alto costo de esos estudios.
Desarrollo histórico de la regulación de los medicamentos genéricos en México
Cuadro Básico de Medicamentos del Sector Salud
En la década de los años setenta García Hernández (Domínguez Pérez, Pérez Rul & Reséndiz Romero, 2008) señaló que la dependencia tecnológica y el dominio de la inversión extranjera eran un problema en México que se agravaba por la falta de políticas de Estado en los aspectos científico y tecnológico. En aquel momento la IF nacional se limitaba a producir algunas formas farmacéuticas sencillas como comprimidos y soluciones, así como a acondicionar (empaquetar) insumos para la salud. El proceso de investigación y desarrollo del sector se encontraba reducido a las escasas actividades de investigación realizadas por empresas extranjeras establecidas en el país.
En el DOF (1975) se expidió el primer texto oficial para seleccionar los medicamentos esenciales para el sector público, al cual se le denominó Cuadro Básico de Medicamentos (CBM). Aunque el documento no incluyó la lista de medicamentos, quedó especificado que, una vez formulada ésta, su uso sería obligatorio para las instituciones de salud y el texto se revisaría periódicamente. Sin embargo, la lista se publicó dos años después.
En el DOF (1977) se conformó al fin la Comisión para Elaborar el CBM del Sector Público. La confección del CBM contó con la opinión profesional de más de 150 médicos de la entonces Secretaría de Salubridad y Asistencia (SSA), el Instituto Mexicano del Seguro Social (IMSS), el Instituto de Seguridad y Servicios Sociales de los Trabajadores del Estado (ISSSTE) y la Academia Nacional de Medicina (ANM). Cabe señalar la lamentable ausencia de farmacéuticos y asociaciones profesionales de farmacia en la elaboración del primer CBM del país.
La Comisión elaboró una lista de 432 “medicamentos genéricos” y 622 presentaciones farmacéuticas distribuidos en 24 grupos, tomando como base las listas disponibles en las instituciones de salud pública del país (Domínguez Pérez, Pérez Rul & Reséndiz Romero, 2008). El CBM constituyó un hito en el desarrollo de la legislación farmacéutica mexicana, debido a que generó un primer punto de acuerdo para las instituciones de salud y constituyó un marco de referencia con base en el cual orientar la producción de medicamentos del país. Asimismo, en años posteriores, el CBM permitiría la compra consolidada de los medicamentos, práctica que hoy en día es utilizada para eficientar recursos mediante la obtención de descuentos por volumen en la compra de insumos para el sector salud.
En el DOF (1983) se redefinieron las atribuciones de la Comisión en la elaboración y revisión del Cuadro Básico de Insumos del Sector Salud (CBISS), como ahora se denominaría al CBM y la divulgación de la información científica pertinente entre las instituciones del país. Este hecho reforzó la puesta en marcha de campañas dirigidas a los médicos para difundir la relación entre las marcas comerciales y las claves correspondientes del CBM del sector público. De esta forma se homologó el CBM en todo el país, permitiendo a las instituciones de salud pública orientar sus compras con base en un mismo criterio que respondía a las necesidades del sector salud.
La Ley General de Salud (LGS), publicada en 1984, estableció un nuevo CBM atendiendo los cuatro criterios de inclusión y exclusión propuestos por la OMS (Domínguez Pérez, Pérez Rul & Reséndiz Romero, 2008) de 1977 (OMS, 2010):
“Selección de los medicamentos sobre la base de la mayor eficiencia con el menor riesgo.
Eliminación de medicamentos cuya calidad haya sido superada e inclusión de nuevos productos de valor terapéutico demostrado.
Supresión de duplicaciones de medicamentos con la misma acción farmacológica.
Exclusión de combinaciones infundadas de medicamentos.”
El hecho de que el CBM se insertara en la LGS ciertamente elevó la jerarquía normativa de este documento, en tanto que esta última es el máximo instrumento legal especializado en materia de salud.
En el año de 1998 (Domínguez Pérez, Pérez Rul & Reséndiz Romero, 2008) se publicaron dos textos legales de gran importancia: el Reglamento de Insumos para la Salud (RIS) y el Catálogo de Medicamentos Genéricos Intercambiables (CMGI). Este último organizó a los medicamentos en 23 grupos terapéuticos e incluyó 74 genéricos intercambiables (GI) repartidos entre esos grupos (Leyva, 1999). El CMGI formaría, junto con el CBM, un recurso importante para la administración de los insumos para la salud en hospitales del sector público.
A diferencia del CBM “Listado de los medicamentos mínimos indispensables con los que deben contar las dependencias que prestan servicios de salud” de 1977, el CMGI era un listado que incluía únicamente aquellos medicamentos autorizados como genéricos intercambiables. Mientras el primero mostraba las sustancias activas, vía de administración, efectos terapéuticos, reacciones adversas y dosis para la administración de medicamentos de patente y genéricos; el segundo incluía exclusivamente medicamentos genéricos autorizados, indicando su forma farmacéutica, presentación y laboratorio productor. En otras palabras el CBM era el universo que comprendía todos los medicamentos incluidos en el CMGI, condición que no se cumplía a la inversa.
La reforma al RIS del 2008 cesó la publicación periódica del CMGI. El último ejemplar vio la luz el 24 de febrero de 2010. Ello se debió a que, partir del año 2008, todos los medicamentos genéricos quedaron obligados a demostrar su intercambiabilidad en función del Acuerdo por el que se adiciona y modifica la relación de especialidades susceptibles de incorporarse al Catálogo de Medicamentos (DOF, 2008) que establece que es un Comité de Expertos del Consejo de Salubridad quien determina el tipo de prueba para documentar la intercambiabilidad.
La Norma Oficial Mexicana NOM-177-SSA1 y los medicamentos genéricos
El 26 de enero de 1999, se publicó en el DOF el proyecto de la ya mencionada NOM-SSA1-177-1998 Que establece las pruebas y procedimientos para demostrar que un medicamento es intercambiable. Requisitos a que deben sujetarse los terceros autorizados que realicen las pruebas, la cual representó el primer acto normativo para la producción de medicamentos genéricos a nivel nacional. La norma introdujo dos conceptos importantes: “equivalentes farmacéuticos” y “productos bioequivalentes”. Los primeros se referían a “… medicamentos que contienen la misma cantidad de la misma sustancia o sustancias activas, en la misma forma farmacéutica, que cumplen con las especificaciones de la FEUM”, y los segundos eran definidos como “Equivalentes farmacéuticos en los cuales no se observa diferencia significativa en la velocidad y cantidad absorbida del fármaco, cuando son administrados ya sea en dosis única o dosis múltiple bajo condiciones experimentales similares”. Es decir los medicamentos debían ser equivalentes farmacéuticos en primera estancia para ser un producto bioequivalente.
En el DOF (1999) también se introdujo un tercer término, el de “medicamento genérico intercambiable”, entendido como la “Especialidad farmacéutica con el mismo fármaco o sustancia activa y forma farmacéutica, con igual concentración o potencia, que utiliza la misma vía de administración y con especificaciones farmacopeicas iguales o comparables, que después de cumplir con las pruebas reglamentarias requeridas, ha comprobado que sus perfiles de disolución o su biodisponibilidad u otros parámetros, según sea el caso, son equivalentes a las del medicamento innovador o producto de referencia, y que se encuentra registrado en el Catálogo de Medicamentos Genéricos Intercambiables, y se identifica con su denominación genérica”. En otras palabras, los medicamentos genéricos intercambiables debían ser productos bioequivalentes.
Un aspecto importante (DOF, 1999) es que estableció el cálculo matemático de un factor de similitud denominado f2 y obtenido a partir de los resultados de los perfiles de disolución (es decir, la prueba in vitro) de los medicamentos de referencia y genérico. El resultado del f2 era satisfactorio si se encontraba entre 50 y 100. De igual manera el 19 de marzo de 1998 se publicó en el DOF el Acuerdo por el que se relacionan las especialidades farmacéuticas susceptibles de incorporarse al Catálogo de Medicamentos Genéricos Intercambiables y se determinan las pruebas que deberán aplicárseles, donde se establecieron las pruebas de intercambiabilidad necesarias para que un medicamento pudiera entrar al CMGI (DOF, 1998b).
Transcurrieron 15 años para que dicha norma fuera modificada. El 6 de mayo del 2013 se promulgó la NOM-177-SSA1-2013 (DOF, 2013a), Que establece las pruebas para demostrar que un medicamento es intercambiable. Requisitos a que deben sujetarse los Terceros Autorizados que realicen las pruebas de intercambiabilidad. Requisitos para realizar estudios de biocomparabilidad. Requisitos a que deben sujetarse los Terceros Autorizados, Centros o Instituciones Hospitalarias que realicen pruebas de biocomparabilidad. Tanto las versiones de 1998 como 2013 de la NOM-177-SSA1 establecen sus límites mediante el uso de parámetros farmacocinéticos como el área bajo la curva (ABC) y la concentración plasmática máxima (Cpmáx). Estos parámetros se obtienen mediante el monitoreo de la concentración del fármaco en la sangre de los individuos, por lo que implican el uso de modelos clínicos humanos.
Las principales diferencias con su predecesora son que la NOM-177-SSA1-1998 (DOF, 1998b): a) no indica de forma expresa la obligatoriedad de contratar los servicios de evaluación in vivo de los nuevos medicamentos genéricos a un Tercero Autorizado (TA) para la obtención de un registro sanitario (sin embargo, el artículo 166, fracción I del RIS sí los considera obligatorios) (DOF, 1998b); b) incluye exenciones de la prueba in vivo; c) introduce el nuevo concepto de “bioequivalencia”, definido como la “Relación entre dos equivalentes farmacéuticos o alternativas farmacéuticas, cuando al ser administrados bajo condiciones similares producen biodisponibilidades semejantes”; d) establece que una alternativa farmacéutica puede ser bioequivalente con el innovador; e) reconoce al TA como responsable de la validación de los métodos analíticos de la prueba de disolución; f) establece que la prueba de disolución puede omitirse si se realizó la de bioequivalencia (mientras que la versión de 1998 exigía ambos estudios); g) adiciona el método general de análisis (MGA) 521 <Liberación Controlada˃ de la FEUM (DOF, 2013a); h) inclusión de medicamentos biocomparables (que se refieren a los medicamentos biotecnológicos).
La versión del 2013 incluyó la definición de TA y enfatizó la importancia de esta figura que, si bien ya estaba definida en el RIS (DOF, 1998a), se amplió sustancialmente (en negritas la inclusión del 2013) quedando como: “ Persona autorizada por la Secretaría de Salud para realizar con probidad, imparcialidad de manera calificada actividades en apoyo al control sanitario, así como la evaluación técnica de actividades, establecimientos, procedimientos y servicios en el territorio nacional o en el extranjero, además de proporcionar información y realizar estudios respecto del cumplimiento de requisitos establecidos por la propia Secretaría de Salud o en las disposiciones jurídicas aplicables, cuyos informes técnicos podrán auxiliar a la autoridad sanitaria ” (DOF, 2013b). En otras palabras, en la versión 2013 la figura del TA se encuentra ligada al proceso de evaluación de medicamentos genéricos de forma imperante y obligatoria.
Esta versión introdujo una serie de conceptos que su predecesora no contemplaba (si bien algunos de ellos se refieren a los medicamentos biotecnológicos biocomparables), como por ejemplo: “estudios preclínicos” y “estudios clínicos”, “control de calidad”, “comité de ética e investigación” y “consentimiento informado”. Asimismo, por vez primera se definió la “bioequivalencia” o “… relación entre dos equivalentes farmacéuticos o alternativas farmacéuticas cuando al ser administrados bajo condiciones similares producen biodisponibilidades semejantes” (DOF, 2013a). Cabe resaltar que los medicamentos genéricos debían cumplir en una primera instancia con el Acuerdo por el que se adiciona y modifica la relación de especialidades farmacéuticas susceptibles a incorporarse al Catálogo de Medicamentos Genéricos (DOF, 2008) y a partir del 2015 deben cumplir con el Acuerdo que determina el tipo de prueba para demostrar intercambiabilidad de medicamentos genéricos (DOF, 2015). Así, la norma 2013 mantiene a la intercambiabilidad como la determinación de manera científica de la equivalencia entre medicamentos (uno de referencia o innovador y uno de prueba) mediante pruebas in vivo (biodisponibilidad y bioequivalencia) o in vitro (perfiles de disolución), según aplique. Para el caso de las pruebas in vitro, esta versión precisó que si el fármaco se ha disuelto en un 85% en 15 minutos o menos no es necesario calcular el f2, (la versión de 1998 expresaba para ese caso que no era necesaria la caracterización de la curva ascendente, pero no eximía el cálculo del f2).
La NOM (DOF, 2013a) también establece que los medicamentos genéricos deben cumplir con el Acuerdo que determina el tipo de prueba para demostrar intercambiabilidad de medicamentos genéricos (DOF, 1998b; DOF, 2015) en el que se señalan las definiciones explícitas de los tres niveles de evaluación para los medicamentos:
A: No requiere someterse a prueba de disolución o bioequivalencia.
B: Prueba de perfil de disolución.
C: Prueba de bioequivalencia.
La exención de los estudios de bioequivalencia es la posibilidad de reemplazar las pruebas in vivo por ensayos in vitro y es aplicable únicamente con la condición de que: a) el fármaco reúna ciertas características biofarmacéuticas y b) que presente una forma farmacéutica sólida de liberación inmediata (González Casado, 2015). La exención es una alternativa atractiva para el fabricante de medicamentos, puesto que permite acelerar el diseño de la formulación, acortar los tiempos de investigación para obtener antes el registro del medicamento genérico y prescindir de los estudios clínicos y del enorme gasto que éstos representan para un laboratorio. El acuerdo antes mencionado especifica que los siguientes medicamentos no deben someterse a pruebas de bioequivalencia o disolución (DOF, 2015):
Las soluciones acuosas para uso parenteral;
Las soluciones orales exentas de excipientes conocidos que modifiquen los parámetros farmacocinéticos;
Los gases (medicinales);
Los medicamentos tópicos de uso no sistémico y cuya absorción no represente riesgo;
Los medicamentos para inhalación en solución acuosa, y
Los medicamentos para inhalación en suspensión y cuyo tamaño de partícula sea demostradamente igual al del innovador.
En la NOM-177-SSA1-2013 (DOF, 2013a) que permite la exención de los estudios de bioequivalencia a las concentraciones bajas de un medicamento, cuando éste se presenta a diferentes concentraciones, siempre y cuando estas concentraciones estén consideradas en el medicamento de referencia; es decir los resultados de la concentración máxima son extrapolables a concentraciones más bajas. Asimismo, debe mantenerse la forma farmacéutica y existir una proporcionalidad en el contenido del (os) fármaco(s) y aditivo(s) en la fórmula cuali-cuantitativa con respecto al medicamento que fue declarado bioequivalente bajo los siguientes criterios (DOF, 2013a):
El perfil de disolución del medicamento evaluado es similar al de referencia
El o los fármaco(os) y excipiente(s) se encuentran en la misma proporción entre las diferentes concentraciones.
El o los fármaco(os) y aditivo(s) no se encuentran en la misma proporción entre las diferentes dosis, pero la relación de ingredientes activos y el peso total de la forma varía solamente hasta un 10%.
Para fármacos de alta potencia donde la dosis es menor a 10 mg, y el peso total de la forma farmacéutica es igual en todas las dosis, y existe proporcionalidad siempre y cuando el peso total de las forma no varía más de un 10%.
Para el caso de formas farmacéuticas de liberación inmediata, los componentes de la cubierta, colorantes, saborizantes y cápsula no deben ser considerados para el cálculo de proporcionalidad.
El medicamento a exentar presenta una farmacocinética lineal a las concentraciones solicitadas.
El proceso de fabricación está validado.
En relación a la bioexención utilizando el SCB existen en el mundo, discrepancias respecto a los criterios específicos que deben cubrirse para realizar una exención de estudios in vivo. La OMS, 2006, permite la exención de estudios in vivo de las clases I del SCB, y bajo condiciones especiales y más estrictas permite la exención de algunos medicamentos de las clases II y III del SBC que cumplen con criterios adicionales (OMS, 2006; Organización Panamericana de la Salud, 2011), mientras que la FDA (Food and Drug Administration, 2015) mantiene una postura más conservadora, permitiendo la exención para los fármacos pertenecientes a la clases I y III.
Ambas organizaciones concuerdan en que los fármacos clase I son los ideales para presentar exención y los de la clase IV no pueden evitar realizar las pruebas in vivo.
Volviendo a la regulación imperante en nuestro país, aparte de los criterios de exención de las pruebas in vivo, el cambio más significativo en la versión 2013 (DOF, 2013a) es la definición de los medicamentos genéricos, como: “…las especialidades farmacéuticas que cumplen con las pruebas de intercambiabilidad señaladas por el Consejo de Salubridad General ”. Es decir que esas pruebas se indican en el Acuerdo que determina las pruebas de intercambiabilidad publicado en el 2008 (DOF, 2008), modificado en 2015 con el nombre de Acuerdo que determina el tipo de prueba para demostrar intercambiabilidad de medicamentos genéricos (DOF, 2015).
Con estas modificaciones la Secretaría de Salud (SSA) abona a la garantía de que los medicamentos genéricos - cuyo bajo costo los proyecta como la alternativa de elección para el tratamiento de las enfermedades tanto en los sistemas de salud público como privado - son eficaces y seguros. Dichas modificaciones comprendieron la inclusión del concepto de bioequivalencia y la actualización de la definición de genérico, ligándola directamente con el Acuerdo 2008 actualizado en 2015. En cuanto al cálculo de f2, cuando el coeficiente de variación (CV) del perfil de disolución sea mayor a 20% en la primera muestra o mayor a 10% en las muestras subsecuentes, se permite un procedimiento denominado de región de certeza multivariado independiente de modelo, que es un tratamiento estadístico alterno que permite verificar la similitud (DOF, 2013a). Dicho modelo está descrito en la Guía para la Industria Pruebas de disolución de formas de dosificación oral sólidas de liberación inmediata (FDA, 1997).
A raíz de la actualización de la NOM-177-SSA1-2013 (DOF, 2013a), el Consejo de Salubridad General ha publicado varias guías obligatorias cuya vigilancia corresponde a la interpretación de esta norma; como la Guía de estudios de bioequivalencia de medicamentos sólidos orales de liberación modificada (Consejo de Salubridad General, 2016a) y la Guía para la evaluación de la intercambiabilidad de medicamentos que contienen fármacos de estrecho margen terapéutico y dosis crítica (Consejo de Salubridad General, 2016b). La vigilancia en el cumplimiento de estas guías corresponde a la Comisión Federal para la Protección contra Riesgos Sanitarios (COFEPRIS) de la Secretaría de Salud.
Actualmente el artículo 225 de la LGS (Ley General de Salud, 1984) señala que los medicamentos pueden ser identificados por sus denominaciones genérico y distintiva, sin embargo, solo la genérica será obligatoria; y el artículo 31, fracción I del RIS en (DOF, 2016) señala que: “En el caso de (la prescripción) de medicamentos genéricos; (el emisor) deberá anotar la definición genérica y si lo desea, podrá indicar la denominación distintiva de su preferencia.” El Suplemento de la FEUM 5ta edición 2014 (Comisión Permanente de la FEUM, 2014b), indica que para la prescripción en las instituciones públicas se utilizarán las denominaciones genéricas de los medicamentos del CBM, y sólo por excepción y autorización podrá prescribir otro medicamento.
De esta forma hoy en día, la población puede identificar a los medicamentos tanto por su nombre distintivo como genérico, y con este último puede seleccionar la mejor opción disponible en las farmacias.
Cabe mencionar que, a pesar de la gran diversidad de medicamentos genéricos a nivel nacional, hoy en día el consumidor final encuentra acotada su capacidad de elección por los convenios entre las farmacias y la IF, que limitan las marcas de medicamentos genéricos disponibles en una sola farmacia; es decir que la elección final de un genérico sobre otro dependerá en primera instancia del sitio en el que se expenda (o, en el mejor de los casos, se dispense) el producto.
Hasta aquí hemos descrito los acontecimientos históricos relativos a la legislación sanitaria en materia de medicamentos genéricos en México. La Figura 4 muestra la línea del tiempo con los hitos que han marcado la regulación de estos insumos para la salud.

Conclusiones
México es el segundo mercado más grande de AL y el principal exportador de medicamentos en la región. No obstante, en el balance comercial de este sector las importaciones de materias primas y medicamentos superan las exportaciones, situación que muestra que el país aún no es capaz de mantenerse así mismo en el sensible rubro de la producción de medicamentos.
México está considerado como un mercado emergente; esta condición puede ser aprovechada para atraer capital extranjero e impulsar la investigación nacional en el sector farmacéutico.
La IF establecida en el país se encuentra concentrada en la región centro, con excepción de Jalisco y algunas cuantas plantas aisladas ubicadas en Celaya y Saltillo, por ejemplo. Una descentralización podría fomentar la creación de empleos en otras regiones de la república, pero requiere cuantiosas inversiones. El sector farmacéutico ha aumentado la contratación de profesionales universitarios y posgraduados; este indicador acusa el interés que existe en las actividades de I&D.
Aunque los medicamentos con el mismo principio activo en la misma cantidad y forma farmacéutica se han vendido en México desde siempre, los genéricos surgieron en 1998. El sistema legal relativo a ellos ha evolucionado significativamente desde 1977, año en que se publicó el primer CBM y en particular en los últimos cinco años se han actualizado varias de las NOM relevantes para el sector farmacéutico como la 059, la 220, la 164 o la 241, entre otras. En NOM-177-SSA1-1998 (DOF, 1999), publicada 21 años después del primer CBM, regularía la producción de medicamentos genéricos. Estas dos décadas fueron suficientes para que en la población mexicana se arraigara un concepto equívoco del papel de los medicamentos genéricos, debido a que el término “similar” se acuñó en el mercado nacional antes que la propia norma técnica los nombrara y definiera; además hubo carencias en la estrategia de comunicación oficial relativa a los medicamentos genéricos y permisión de la publicidad con términos engañosos en los medios de comunicación masivos. El divorcio entre los términos usados vulgarmente y los expresados en la legislación sanitaria no ha podido superarse del todo.
Entre 1998 y el 2010 se publicaron periódicamente actualizaciones del CMGI y a partir del 2008 se publicó en el DOF un Acuerdo por el que se adiciona y modifica la relación de especialidades farmacéuticas susceptibles a incorporarse al catálogo de medicamentos genéricos, el cual determina el tipo de prueba de intercambiabilidad para considerar medicamentos genéricos. Este acuerdo sufrió su más importante modificación en julio de 2015. Con la eliminación del CMGI los genéricos entraron al mercado, lo cual facilitó la disponibilidad de dichos medicamentos entre la población.
En el 2013, con la publicación de la NOM-177-SSA1-2013 (DOF, 2013), México se adhirió a las sugerencias de la OMS de 2006 en materia de medicamentos genéricos. Ello beneficiaría la salud de los pacientes mexicanos. Tanto la LGS y el RIS como esta NOM favorecen la inclusión dinámica de nuevos medicamentos genéricos al mercado nacional.
El SCB ha sido un importante referente para la exención de los estudios in vivo para las formas farmacéuticas orales de liberación inmediata, pero a pesar de su sustento en bases científicas, es interpretado a discreción por los diferentes organismos de relevancia internacional. Sin embargo, nuestro país no ha publicado ninguna guía de referencia con base en el SCB.
Con todo, el rigor científico expresado en la legislación mexicana vigente ha permitido que el sector farmacéutico de medicamentos genéricos se haya consolidado y que se pueda confiar en los medicamentos producidos en este país. La regulación clara sobre medicamentos genéricos es necesaria para que el Estado garantice el acceso a los medicamentos y con ello haga cumplir uno de los máximos principios constitucionales: el derecho a la salud de los mexicanos.

