











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink1. Introducción y antecedentes
El genoma de un organismo se puede considerar como el conjunto de instrucciones, codificadas en una secuencia de nucleótidos (ACGT), que contiene toda la información necesaria para formar un organismo y heredar estas características a sus descendientes [1].
La obtención del genoma de un organismo se realiza mediante tecnologías de secuenciación. En particular las tecnologías de secuenciación de nueva generación (NGS), que generan millones de lecturas cortas y un gran volumen de datos, han revolucionado la biología molecular impactando en áreas como la académica, médica, farmacéutica, biotecnológica, agroquímica y en la industria alimentaria, entre otras [2].
Debido a las limitaciones de los equipos NGS, el primer paso en el proceso de secuenciación consiste en fragmentar la cadena de ácido desoxirribonucleico (ADN) de manera aleatoria, y por ende no se cuenta con información de la posición relativa de cada fragmento [3]. Para poder recuperar la información contenida en la cadena original se requiere entonces un proceso de reconstrucción conocido como ensamblaje.
Existen dos formas primordiales de ensamblaje: la reconstrucción de la cadena alineándola a un genoma de referencia, conocida como mapeo; y la reconstrucción a partir únicamente de los fragmentos, conocida como ensamblaje de novo. El ensamblado de novo de lecturas de NGS es complejo debido a varios factores, entre ellos, el elevado número de secuencias y su tamaño reducido, así como a los errores de secuenciación y la repetición de cadenas, entre otros [4].
Si bien el conjunto de instrucciones necesarias para el funcionamiento y formación de un organismo se encuentra contenido en su genoma (ADN), las células requieren de moléculas informativas intermediarias, ácidos ribonucleicos (ARN), para dirigir la producción de moléculas de trabajo (proteínas) en un proceso conocido como traducción. El proceso de envío de instrucciones contenidas en el ADN por medio de ARNs se conoce como transcripción, ya que dichas instrucciones no son copias exactas de los segmentos de ADN de las cuales provienen; dichas instrucciones se conocen como transcritos [5]. El término transcriptoma por consiguiente, se refiere al conjunto de ARNs transcritos que representan genes expresándose en un momento dado, proporcionando el estado biológico de la célula [1].
De manera similar a como se realiza la secuenciación de un genoma, se puede secuenciar un transcriptoma.
Pero, un transcriptoma es más complejo; éste contiene miles de transcritos con distinto nivel de abundancia, además una sola secuencia del genoma (gen) puede transcribirse en varias secuencias de transcriptoma (isoformas), debido a que sus fragmentos se pueden alinear de diversas formas (splicing alternativo). En consecuencia, cuando se ensamblan las lecturas obtenidas de la secuenciación de un transcriptoma, la fase inicial del ensamblaje realiza una agrupación de las secuencias pertenecientes al mismo gen (clustering).
Sin embargo, puede incurrirse en el caso de que los transcritos sean agrupados erróneamente en un mismo cluster. Asimismo, otro problema consiste en agrupar genes muy similares (parálogos). Reconstruir todos los transcritos e isoformas que se encuentran expresados, es decir ensamblarlos, ha requerido del desarrollo de nuevos algoritmos computacionales capaces de procesar la gran cantidad de secuencias cortas generadas por las tecnologías NGS [6]. Los algoritmos basados en grafos De Bruijin (DBG) o ensambladores Eulerianos han demostrado ser los más aptos para estas tecnologías [7].
Los algoritmos que usan DBGs se basan en extraer primeramente fragmentos únicos de longitud k (k-meros) a partir de las secuencias originales; posteriormente se conforman los nodos del grafo con subsecuencias de longitud k-1 provenientes de los k-meros. A continuación, conectan los nodos considerando prefijos y sufijos (k-1-meros) a través de grafos dirigidos. Por último, resuelven la trayectoria a través de un ciclo Euleriano para formar una supercuerda que visite los nodos solo una vez y termine donde empezó [8]. No obstante, el algoritmo presupone condiciones tales como que no existan errores en los nodos, que se encuentre completo el alfabeto de k-meros, la existencia de un camino optimo que genere 1 sola supersecuencia y otros que no se cumplen en el proceso de secuenciación. Por ejemplo, la lectura incorrecta de bases puede generar k-meros incorrectos, en tanto que la aparición de secuencias repetidas, consecuencia del splicing alternativo, implica que más de una supercuerda sea posible [7].
Estos y otros factores relacionados con el proceso biológico, la toma de la muestra, la tecnología de secuenciación, errores de secuenciación, preprocesamiento con base en la calidad de las lecturas obtenidas, la selección del programa para ensamblar e inclusive los parámetros de ensamblaje influyen en el ensamblaje obtenido. Todos estos factores han sido estudiados en otras investigaciones [9-11]. Sin embargo, un tema que no se ha abordado con suficiente profundidad es la influencia que pudiese tener la disposición y asignación de recursos computacionales para una tarea de ensamblaje de novo.
Es común que un proyecto que involucre ensamblaje de novo esté enfocado en los aspectos de laboratorio en lugar de los recursos computacionales requeridos. Adicionalmente, una de las principales limitantes para una investigación en la implementación de un ensamblador de novo es la demanda computacional de estos programas [12]. Típicamente, un ensamblador de novo de vanguardia corre en plataformas multinúcleo, con bastante memoria RAM, con alta capacidad de almacenamiento y sistemas operativos Linux; frecuentemente sobre equipamiento de Cómputo de Alto Rendimiento (High Performance Computing, HPC), no disponibles en muchas instituciones.
Los reportes actuales sobre ensamblaje de novo típicamente muestran los efectos que la cantidad memoria y núcleos tienen sobre los tiempos de procesamiento o sobre la viabilidad de la plataforma para la ejecución del proceso [12-14]. No obstante, apenas se tienen precedentes de la existencia de una ligera variación en la salida del ensamblaje, resultado de correr el proceso en distintas ocasiones o en hardware distinto [15]. Asimismo, se ha reportado que existe aleatoriedad en los resultados (de ensamblaje) debido al uso de multi-threading en combinación con la utilización de estructuras probabilísticas de datos [16].
En ambos trabajos no cuantificaron experimentalmente dicha variabilidad y aleatoriedad en los ensamblajes. Dadas las condiciones típicas de ensamblaje y los pocos precedentes de los efectos computacionales en estos procesos, el efecto que un equipo de cómputo tiene en la obtención de ensamblajes de novo no es tomado en cuenta y no se aprovecha en favor de la prospección y generación de información de transcriptomas de especies poco estudiadas.
En este trabajo se considera que la influencia del equipo de cómputo y la asignación de recursos computacionales es relevante en el caso de ensamblaje de novo de transcriptoma, dado que el proceso de clustering en las primeras etapas a partir de las cuales se determinan las isoformas, depende de la disponibilidad inicial de secuencias y la incorporación sucesiva de secuencias candidatas al cluster. La distribución inicial de secuencias en las localidades de memoria disponibles para cada procesador influirá, por ende, en la formación de estos clusters y en el resultado final del ensamblaje.
A partir de esta hipótesis, en este trabajo se explora la influencia que tiene la asignación de los recursos computacionales en el ensamblaje de novo de transcriptoma, en términos de repetitividad de un ensamble bajo las mismas condiciones, entre condiciones distintas, y en términos de calidad al comparar lo obtenido con un transcriptoma de referencia.
2. Metodología
Para evaluar la repetitividad y calidad se realizaron ensamblajes en diferentes plataformas computacionales, utilizando organismos modelo para los cuales, sí existe una referencia contra la cual comparar y determinar la calidad de los transcriptomas ensamblados. Se decidió utilizar el software ensamblador de transcriptomas Trinity (ver. 2.1.1) [17], por ser considerado por la comunidad científica como el ensamblador por defecto para realizar ensamblaje de novo [13].
2.1. Recursos computacionales
Dado que el ensamblaje de novo de transcriptoma demanda el uso de cómputo intensivo, se seleccionó como sistema mínimo una estación de trabajo y dos sistemas de HPC, variando en cada plataforma la asignación de memoria y de núcleos de cómputo, tal como se muestra en la Tabla 1. Todas las plataformas cuentan con sistemas operativos Linux de 64 bits.
Tabla 1 Configuraciones de las plataformas de cómputo para realizar ensamblajes
| Plataforma de cómputo | Parámetros Trinity |
Planificador | |||||
|---|---|---|---|---|---|---|---|
| SLURM | TORQUE | ||||||
| Nombre | Plataforma | Memoria RAM (GB) |
Núcleos | Máxima Memoria | CPU | Núcleos | Nodo/ Núcleos |
| W 1 | Estación de trabajo | 20 | 6 | 20 | 6 | - | - |
| W 2 | 24 | 6 | 24 | 6 | - | - | |
| H 1 | HPC (No Virtual) |
128/nodo | 20/nodo | 24 | 6 | - | 1/6 |
| H 2 | 128/nodo | 20/nodo | 64 | 10 | - | 1/10 | |
| V 1 | HPC (Virtual) |
128/nodo | 24/nodo | 24 | 6 | 6 | - |
| V 2 | 128/nodo | 24/nodo | 64 | 12 | 12 | - | |
“-” equivale a “No aplica”
En la estación de trabajo la memoria RAM se limitó físicamente, en tanto que en las plataformas de HPC, dado que no es posible modificar físicamente sus recursos de hardware, se variaron los recursos asignados al trabajo de ensamblaje a través del planificador correspondiente, en cuanto al número de nodos de cómputo y núcleos por nodo.
En el ensamblador Trinity se asignó el parámetro de memoria y el número de hilos por CPU para corresponder con las configuraciones de hardware. Los demás parámetros fueron especificados a sus valores por defecto.
Se utilizó una estación de trabajo Dell Precision T7500, con un procesador Intel Xeon X5680 3.3 GHz de 6 núcleos, con capacidad en discos duros de 2.5 TB, a la cual se le modificó la memoria RAM (20 GB, 24 GB) de acuerdo a las configuraciones W 1 y W 2, respectivamente (Tabla 1).
Los centros HPC donde se realizaron los procesos de ensamblaje son: Laboratorio Nacional de Supercómputo del Sureste de México (LNS) de la Benemérita Universidad Autónoma de Puebla [18] y el proveedor Penguin Computing, a través de su servicio en la nube Penguin On Demand[19].
El primer recurso de HPC está conformado por la supercomputadora Cuetlaxcoapan del LNS, compuesta de un cluster estándar de cálculo con procesadores Intel Xeon y un cluster con procesadores Intel Xeon Phi Knights Landing. El cluster estándar está compuesto de 228 nodos de cálculo Thin y otros 42 nodos de cálculo más robustos (fat, semi-fat, ultra-fat). Para los procesos de ensamble realizados en este estudio se utilizó el cluster estándar, el cual funciona en modo virtual y donde los nodos de cálculo Thin, tienen 2 procesadores Intel Xeon E5-2680 v3 (Haswell) a 2.5 GHz, con 24 núcleos en total y 128 GB de memoria RAM. Los nodos están intercomunicados con una red Ethernet Gigabit y una red Infiniband FDR a 56 Gbps. Este cluster utiliza el administrador de carga de trabajos SLURM [20], que es libre y puede manejar un cluster Linux de cualquier dimensión. La especificación de los recursos computacionales a utilizar en cada ensamblaje se definió en el Job Script, a través de los parámetros de SLURM:
#SBATCH -n 24 # number of MPI tasks (cores) requested,
#SBATCH --ntasks-per-node=24 # task (cores) per node (maximum 24).
Este ejemplo especifica que se ejecute el trabajo con 24 núcleos (1 nodo Thin) del cluster estándar, donde cada núcleo obtiene 5.3 GB de RAM [18]. Esta plataforma computacional, se utilizó para realizar los ensambles en las configuraciones V 1 y V 2 (Tabla 1), especificando el uso de 1 nodo en el Job Script, pero variando la cantidad de núcleos en concordancia con los parámetros de entrada de Trinity.
El segundo recurso de HPC utilizó el servicio en la nube POD de Penguin Computing con la cola T30, que especifica nodos con un procesador Intel Xeon E5-2660 v3 (Haswell) a 2.6 GHz con 20 núcleos y 128 GB de RAM.
Todos los nodos están intercomunicados con una red Ethernet Gigabit de 10 Gbps y una red Infiniband QDR a 40 Gbps. El servicio utiliza el planificador PBS TORQUE [21] para introducir trabajos al cluster computacional. Sin embargo, es necesario seleccionar una cola del planificador. Cada cola provee diferentes tipos de nodo de cómputo, y por lo tanto tienen diferentes precios.
La especificación de los recursos se definió en el Job Script, a través de los parámetros de TORQUE:
Este ejemplo especifica que se ejecute el trabajo con 1 nodo de 20 núcleos de la cola T30, donde cada núcleo tiene 6.4 GB de RAM [19]. Se utilizó esta plataforma computacional, especificando en el Job Script el uso de 1 nodo, pero variando el número de núcleos en concordancia con los parámetros de entrada de Trinity; estas disposiciones se utilizaron para realizar los ensambles en las configuraciones H 1 y H 2 de la Tabla 1.
2.2. Monitoreo de memoria
Con la finalidad de cuantificar el uso de recursos durante el proceso de ensamblaje se decidió monitorear el proceso mediante el comando top de Linux. Posteriormente, se analizaron los datos con Matlab (r2013a) [22]. El registro generado a partir de un muestreo cada 10 segundos y el registro de tiempos (Trinity.timing) permiten identificar la etapa del proceso de ensamblaje que hace el uso más extensivo de memoria.
2.3. Organismos
Aun cuando el ensamblaje de novo se utiliza principalmente en organismos que no cuentan con un genoma o transcriptoma de referencia, en este estudio se requieren referencias para obtener la calidad de los ensamblajes, por lo que se seleccionaron dos organismos modelo: la Mosca de la Fruta (Drosophila melanogaster) y la Pulga de Agua (Daphnia pulex).
Las lecturas crudas de la Mosca de la Fruta, fueron obtenidas del Sequence Read Archive (SRA) [23] del Centro Nacional de Información Biotecnológica (NCBI), con número de identificación SRR042489, provenientes del proyecto [24]. Las lecturas de la Pulga de Agua fueron descargadas de repositorio ENA [25] perteneciente al Instituto Europeo de Bioinformática, número de identificación SRR2075894, obtenidas en el proyecto [26].
Se analizó la calidad de las lecturas con FastQC [27] y según sus resultados se pre-procesaron los datos con Trimmomatic (versión 0.32) [28].
Los transcriptomas de referencia fueron descargados del repositorio Ensembl [29]. Para la Mosca de la Fruta fue la versión 87, que contiene 30,651 transcritos. Para la Pulga de Agua fue la versión GCA_000187875.1 con 30,590 transcritos.
2.4. Métricas
2.4.1. Repetitividad por plataforma computacional
Dado un conjunto de lecturas de secuenciación L m de una especie m obtenidas de una base de datos pública, sea E (p,m,i) un ensamble (un conjunto constituido por contigs) realizado con configuración de plataforma computacional p, utilizando L m como entrada al proceso de ensamblaje de novo, donde i es el número correspondiente a la repetición del proceso de ensamblaje utilizando las mismas condiciones iniciales, se tiene que:
donde I (p,m,n) representa el conjunto de contigs resultantes de la intersección entre los n ensambles de novo, con las mismas condiciones.
Asimismo, se puede decir que el conjunto de contigs no intersectados
Por lo tanto, el conjunto
De tal manera que la cantidad total de contigs obtenidos por plataforma p para un organismo m en n repeticiones está dada por la unión de sus conjuntos intersectados y no intersectados:
Los análisis de conjuntos intersectados y no intersectados se realizaron con el software de cómputo científico Matlab (r2013a) [22].
La cuantificación de la repetitividad se da con base al porcentaje que representa el subconjunto I
(p,m,n)
del conjunto Ctotal
(p,m,n)
y la variabilidad se da con base al porcentaje que representa el subconjunto
Finalmente, la ganancia por variabilidad entre plataformas se cuantifica tomando en cuenta la relación de la variabilidad máxima de las configuraciones de la estación de trabajo entre la variabilidad máxima de las configuraciones en las plataformas basadas en HPC.
2.4.2. Calidad
Si bien existe variabilidad entre un ensamblaje y otro, aun partiendo de las mismas condiciones iniciales, es necesario considerar si esta variabilidad representa contigs presentes en el transcriptoma del organismo o son artefactos del proceso matemático-computacional. Para identificar la validez de los contigs generados es necesario realizar un proceso de mapeo de los contigs al transcriptoma de referencia, definiendo así la calidad del ensamblaje.
El término calidad de ensamblaje, se refiere a la concordancia entre el ensamblaje y el transcriptoma original del organismo modelo [30] o en este caso, a su referencia codificante más próxima y el conjunto de contigs obtenidos del proceso de ensamblado. En este estudio el objetivo es detectar la calidad de los contigs originados exclusivamente por la variabilidad de cada plataforma de cómputo.
En este contexto, se define como referencia codificante a la mejor aproximación de ensamblado de transcriptoma disponible en cierta fecha o versión, disponible en las bases públicas de los organismos involucrados, ver el sitio de Ensembl [13, 29].
Formalmente, sean los conjuntos I
(p,m,n)
e
donde (c, t) representa un par (contig, transcrito) y Im (p,m) es el conjunto de contigs intersectados generados con la plataforma computacional p, que mapearon en el conjunto referencia 𝑇𝑚 · 𝑇(𝑝,𝑚) es el subconjunto de transcritos de T m a los que mapearon los contigs intersectados del conjunto Im (p,m) . Nótese que, para simplificar, se omitió el subíndice n, así mismo en:
donde (c, t) representa un par (contig, transcrito) e
Ya que pueden existir contigs intersectados y no intersectados que mapean a un transcrito común, se puede realizar la siguiente operación:
donde
donde
Asimismo, se puede realizar la siguiente operación:
donde
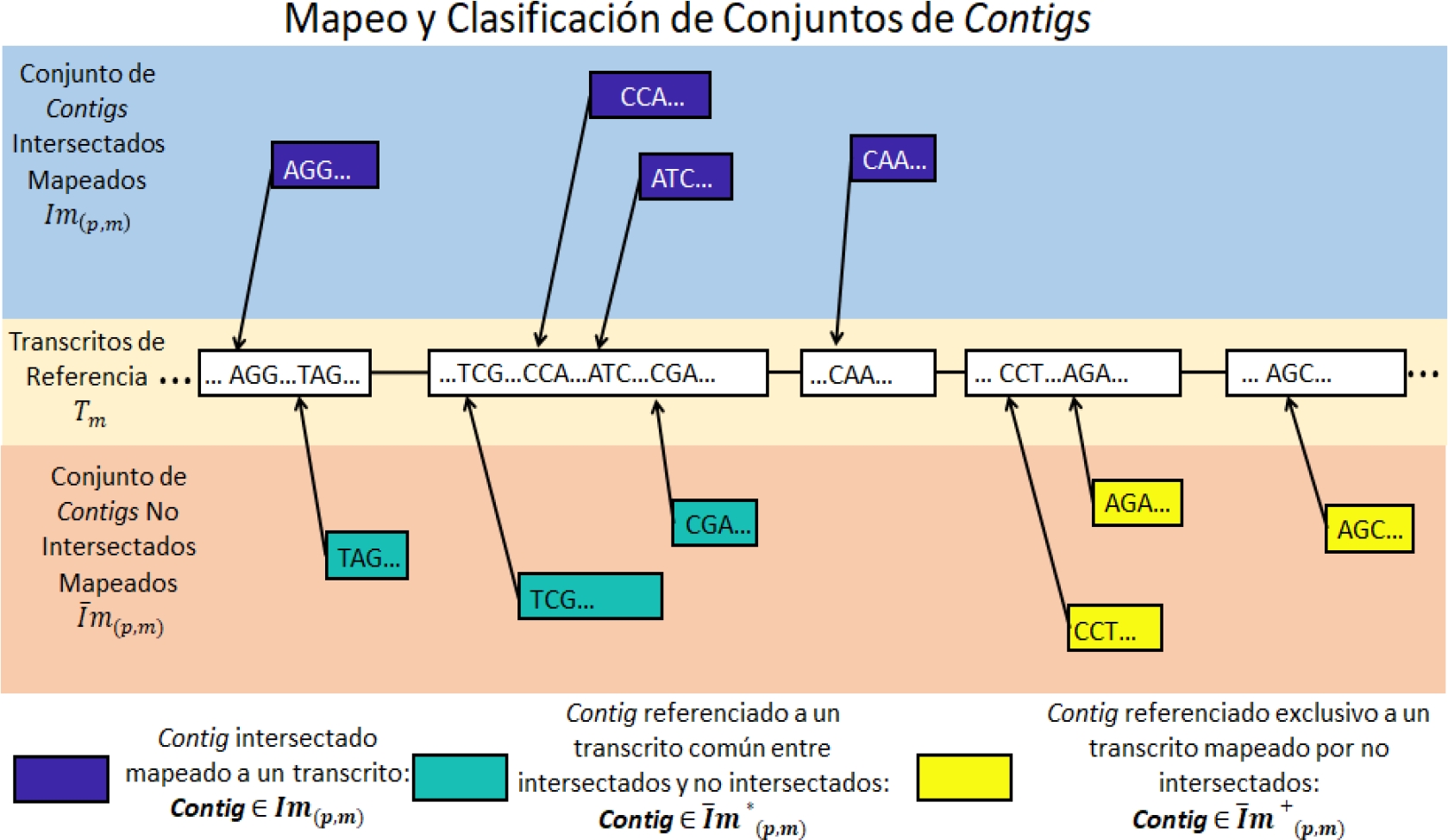
Fig. 1. Mapeo de contigs intersectados y no intersectados, así como su clasificación dependiendo del transcripto al que hayan sido referenciados
Para decidir que contigs se compartían entre ensamblajes se utilizó un criterio estricto de coincidencia única, es decir, ambas cadenas deberían ser exactamente iguales en tamaño y composición. No obstante, los algoritmos de mapeo tienen un criterio más laxo, permitiendo reconocer secciones similares aun cuando no sean las cadenas exactamente iguales. Este criterio permite variar la longitud y composición delcontig.
Debido a este criterio pudiese haber contigs que siendo ligeramente distintos mapean a la misma porción del transcriptoma.
Esta situación puede ocurrir para ambos conjuntos (comunes y no compartidos) y entre conjuntos. Por ello, la evaluación de calidad considera como información válida originada por variabilidad de plataforma a todos aquellos contigs contenidos en el subconjunto
Asimismo, considera como información nueva originada por la variabilidad de plataforma solo a los contigs mapeados exclusivos al conjunto no intersectado
La ganancia en calidad se cuantifica tomando en cuenta la relación del número máximo de contigs representados por el conjunto
De la misma forma, la ganancia en información nueva se cuantifica tomando en cuenta la relación del número máximo de contigs representados por el conjunto
Cabe hacer notar que los parámetros del software BLAST fueron establecidos de tal manera que se obtuviese un hit (alineamiento positivo) por secuencia de entrada, alta similitud entre secuencias alineadas, pero con bajos valores de expectación. Umbrales utilizados: valor de expectación e-value 1x10-9; porcentaje de identidad: 95%; alineamientos máximos por secuencia de entrada max_hps: 1; cantidad de secuencias alineadas max_target_seqs:1; cantidad de núcleos: 1.
3. Resultados
3.1. Datos de entrada
Las estrategias de pre procesamiento para los datos de secuenciación se realizaron basadas en los correspondientes reportes de calidad de las lecturas para cada organismo. Para Mosca de la Fruta fue: corte de primeras 10 bases, remoción de adaptadores en modo palíndromo, l > 32. En los datos de Pulga de Agua: corte de primeras 10 bases, remoción de adaptadores en modo palíndromo, Q min 25, l > 32; posterior al preprocesamiento los datos seguían conteniendo secuencias sobrerrepresentadas, ribosómicas según la búsqueda en la base de datos del NCBI [32], consecuentemente se realizó un segundo pre-procesamiento para remover dichas secuencias.
Los datos de entrada pre-procesados fueron 7,564,138 y 7,168,393 lecturas pareadas con longitudes variables de 32-66 bases para Mosca de la Fruta y Pulga de Agua, respectivamente.
3.2 Contigs ensamblados
La Tabla 2 muestra el promedio de contigs generados después de 5 repeticiones del ensamblaje efectuados en cada configuración. De los promedios y desviaciones se puede apreciar que el número de contigs generados en cada repetición es muy parecido.
Tabla 2 Cantidad de Contigs ensamblados por plataforma computacional
| Mosca de la Fruta | Pulga de Agua | |||
|---|---|---|---|---|
| Plataforma Computacional |
Promedio | Desviación Estándar |
Promedio | Desviación Estándar |
| W 1 | 25,994.80 | 6.72 | 53,280.4 | 37.63 |
| W 2 | 25,988.80 | 5.81 | 53,276.8 | 39.93 |
| H 1 | 25,981.60 | 3.44 | 53,220.8 | 20.32 |
| H 2 | 25,984.40 | 5.68 | 53,247.8 | 15.08 |
| V 1 | 25,988.40 | 3.65 | 53,321.6 | 23.06 |
| V 2 | 25,989.40 | 2.30 | 53,320.0 | 23.98 |
3.3. Uso de memoria
El ensamblador Trinity consta de tres módulos: Inchworm, donde realiza la agrupación inicial de lecturas pertenecientes al mismo gen (clustering) y una construcción extendida de secuencias con base en dichos clusters; Chrysalis:
− Es el módulo donde construye los grafos De Bruijn con base en los clusters de lecturas y los contigs extendidos, para finalmente pasar al módulo; Butterfly, donde resuelve ambigüedades en los grafos con base en la cantidad de lecturas que respaldan la trayectoria de análisis [15].
El módulo Inchworm es el primero en ser ejecutado. Posteriormente, los módulos Chrysalys y Butterfly se ejecutan de forma alterna.
La Figura 2 muestra el uso de memoria RAM durante los procesos de ensamblaje de novo de transcriptoma para Mosca de la Fruta y Pulga de Agua en las plataformas H 2 y W 2. Se delimitó con una línea vertical intermitente la duración del módulo Inchworm.
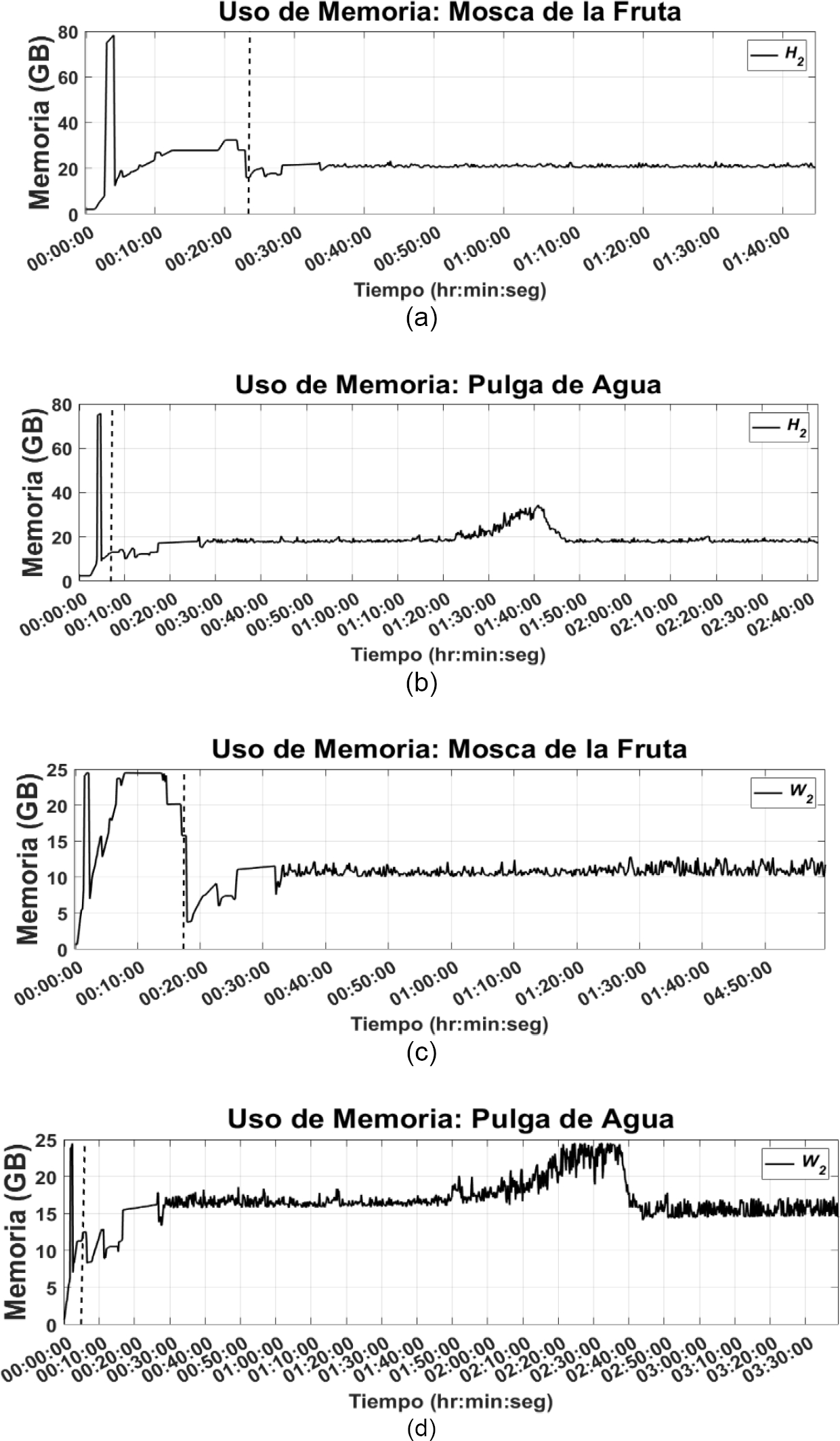
Fig. 2. Utilización de memoria RAM durante el ensamblaje en la plataforma H 2 de Mosca de la Fruta en las configuraciones (a) y Pulga de Agua (b); ensamblaje en la plataforma W 2 de Mosca de la Fruta (c) y Pulga de Agua (d). Línea vertical intermitente indica fin del módulo Inchworm
Nótese que el manejo de los millones de secuencias de entrada se ven reflejadas en el manejo de memoria por parte del ensamblador, sobre todo en el primer módulo, Inchworm, donde ambos organismos hicieron uso intensivo de memoria, ~80 GB en la plataforma H 2 (Figura 2 a y b). La duración del primer módulo en las configuraciones H 2 fue de ~24 minutos en procesos de Mosca de la Fruta y ~5 minutos en ensamblajes de Pulga de Agua. Asimismo, se puede observar que el uso de memoria en los módulos posteriores fue mayor en los procesos de Pulga de Agua teniendo un pico de uso en 34.3 GB (Tabla 3). El uso de memoria en los módulos alternados Chrysalis y Butterfly en el caso de Mosca de la Fruta no excedió los 24 GB.
Tabla 3 Utilización máxima de memoria RAM (GB) por plataforma computacional, organismo y módulo de ensamblaje de Trinity
| Mosca de la Fruta | Pulga de Agua | |||
|---|---|---|---|---|
| Plataforma Computacional | Inchworm | Chr/Btf | Inchworm | Chr/Btf |
| W 1 | 20.4 | 9.7 | 20.4 | 20.4 |
| W 2 | 24.4 | 12.8 | 24.5 | 24.5 |
| H 1 | 21.1 | 13.0 | 26.0 | 27.8 |
| H 2 | 78.3 | 23.3 | 75.7 | 34.3 |
Inchworm: Primer módulo del ensamblador. Chr/Btf: Segundo y tercer módulo del ensamblador Trinity, Chrysalis y Butterfly respectivamente
El uso de memoria en las plataformas W 1 y W 2 se vio limitado por la capacidad física de memoria de las plataformas, inclusive los ensamblajes de Mosca de la Fruta tendieron a saturar el primer módulo (Figura 2 c), y se observaron picos máximos de memoria en al menos 2 módulos al procesar la Pulga de Agua (Figura 2 d).
En la Tabla 3 se muestra la utilización máxima de memoria por configuración, especie y módulo de Trinity.
3.4. Repetitividad por intersecciones
Las figuras 3 y 4 muestran el resultado de intersectar los conjuntos generados con las 5 repeticiones del ensamblaje por máquina. Las intersecciones son los contigs comunes a los ensamblajes que contienen exactamente las mismas secuencias. Basta un cambio de base, inserción o perdida entre dos contigs para que se consideren ambos distintos y se envíen al conjunto de no intersectados. Nótese la diferencia de escalas, y que los contigs no intersectados constituyen menos del 5% en el caso de la Mosca de la Fruta y menos del 30% para Pulga de Agua.
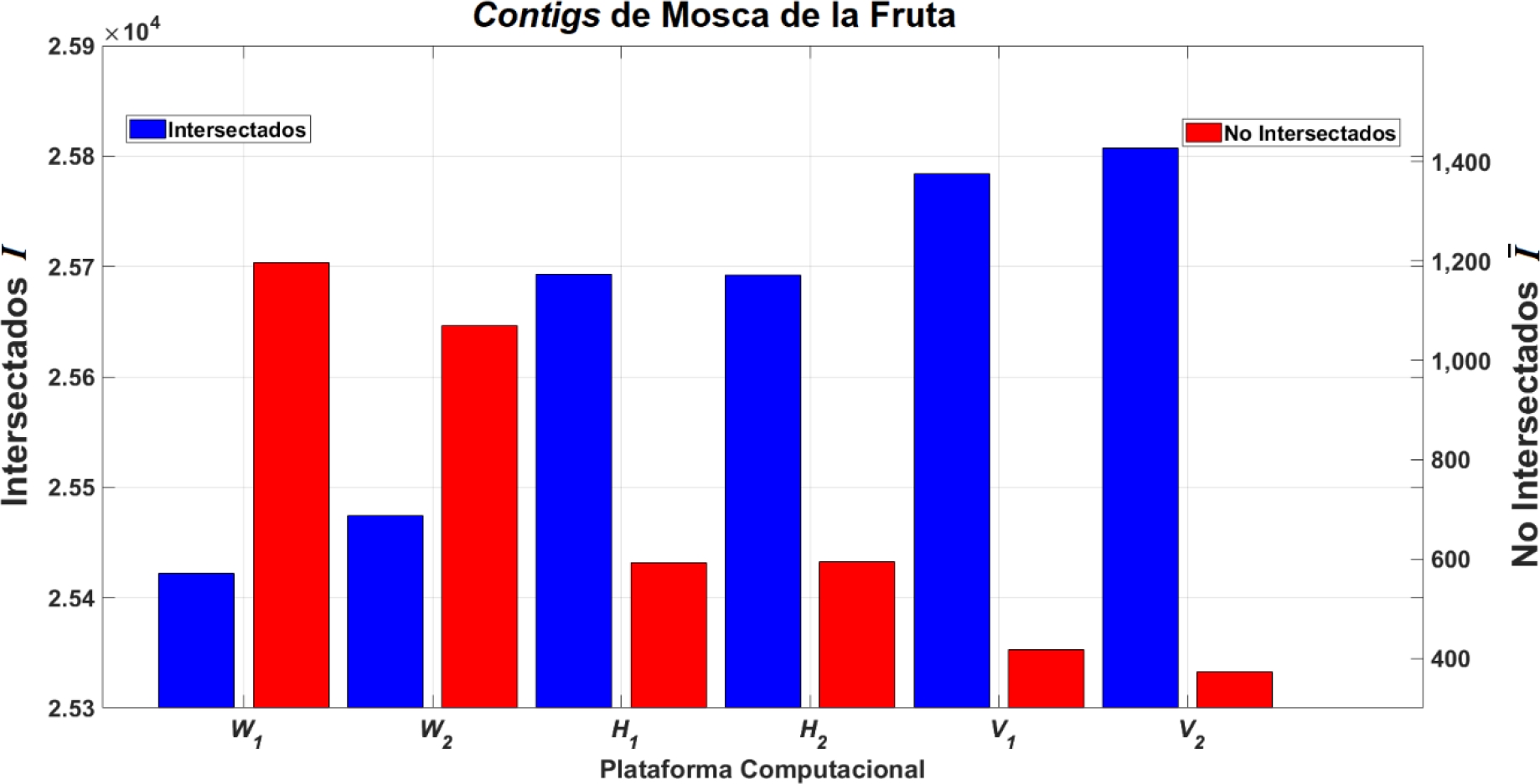
Fig. 3. Comparación de los conjuntos de contigs intersectados I
(p,mosca,5)
y los conjuntos de contigs no intersectados
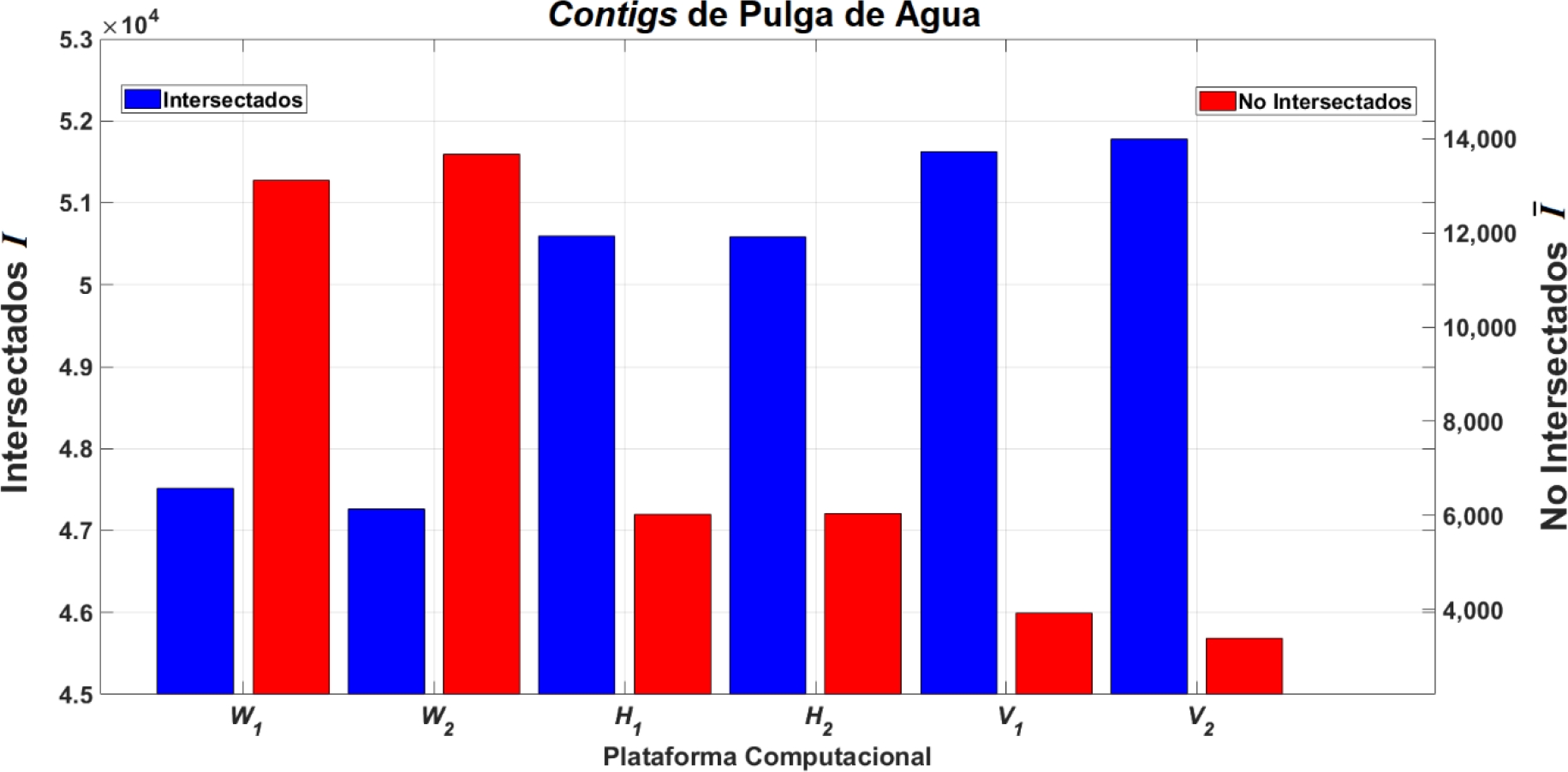
Fig. 4. Comparación de los conjuntos de contigs intersectados I
(p,pulga,5)
y los conjuntos de contigs no intersectados
En la Tabla 4 se muestran los porcentajes de repetividad y variabilidad por plataforma para ambos organismos, encontrándose mayor repetitividad en las plataformas HPC, pero mayor variabilidad en las plataformas con menor memoria. Se observa también que la ganancia máxima por variabilidad de una de las configuraciones basadas en la estación de trabajo, para Mosca de la Fruta es 4.49/2.26 = 1.98 veces más que el máximo de una de las plataformas basadas en HPC; para Pulga de Agua es 22.45/10.68 = 2.10 veces más que el máximo de una de las plataformas basadas en HPC.
Tabla 4 Repetitividad y Variabilidad
| Mosca de la Fruta | |||||
|---|---|---|---|---|---|
| Plataforma Computacional |
|
|
Repetitividad (%) |
|
Variabilidad (%) |
| W 1 | 26,618 | 25,422 | 95.51 | 1,196 | 4.49 |
| W 2 | 26,544 | 25,474 | 95.97 | 1,070 | 4.03 |
| H 1 | 26,286 | 25,693 | 97.74 | 593 | 2.26 |
| H 2 | 26,286 | 25,692 | 97.74 | 594 | 2.26 |
| V 1 | 26,202 | 25,784 | 98.40 | 418 | 1.60 |
| V 2 | 26,180 | 25,807 | 98.58 | 373 | 1.42 |
| Pulga de Agua | |||||
|
Plataforma Computacional |
|
|
Repetitividad (%) |
|
Variabilidad (%) |
| W 1 | 60,632 | 47,510 | 78.36 | 13,122 | 21.64 |
| W 2 | 60,943 | 47,261 | 77.55 | 13,682 | 22.45 |
| H 1 | 56,617 | 50,590 | 89.35 | 6,027 | 10.65 |
| H 2 | 56,630 | 50,580 | 89.32 | 6,050 | 10.68 |
| V 1 | 55,545 | 51,627 | 92.95 | 3,918 | 7.05 |
| V 2 | 55,176 | 51,783 | 93.85 | 3,393 | 6.15 |
Repetitividad: I
(p,m,n)
/ Ctotal
(p,m,n)
. Variabilidad:
3.5. Calidad por mapeos
De igual forma, el mapeo de los contigs de los conjuntos no intersectados por plataforma fue mayor para los conjuntos provenientes de las plataformas con menor memoria (W 1 y W 2).
Las figuras 5 y 6 muestran el mapeo por plataforma computacional para los organismos Mosca de la Fruta y la Pulga de Agua, respectivamente. También, se observan en la Tabla 5 los mapeos de contigs intersectados con respecto a las referencias y los porcentajes que estos representan.

Fig. 5. Comparación de los conjuntos de contigs intersectados mapeados Im
(p,mosca)
al transcriptoma de referencia de la Mosca de la Fruta T
mosca
, contigs no intersectados mapeados exclusivos
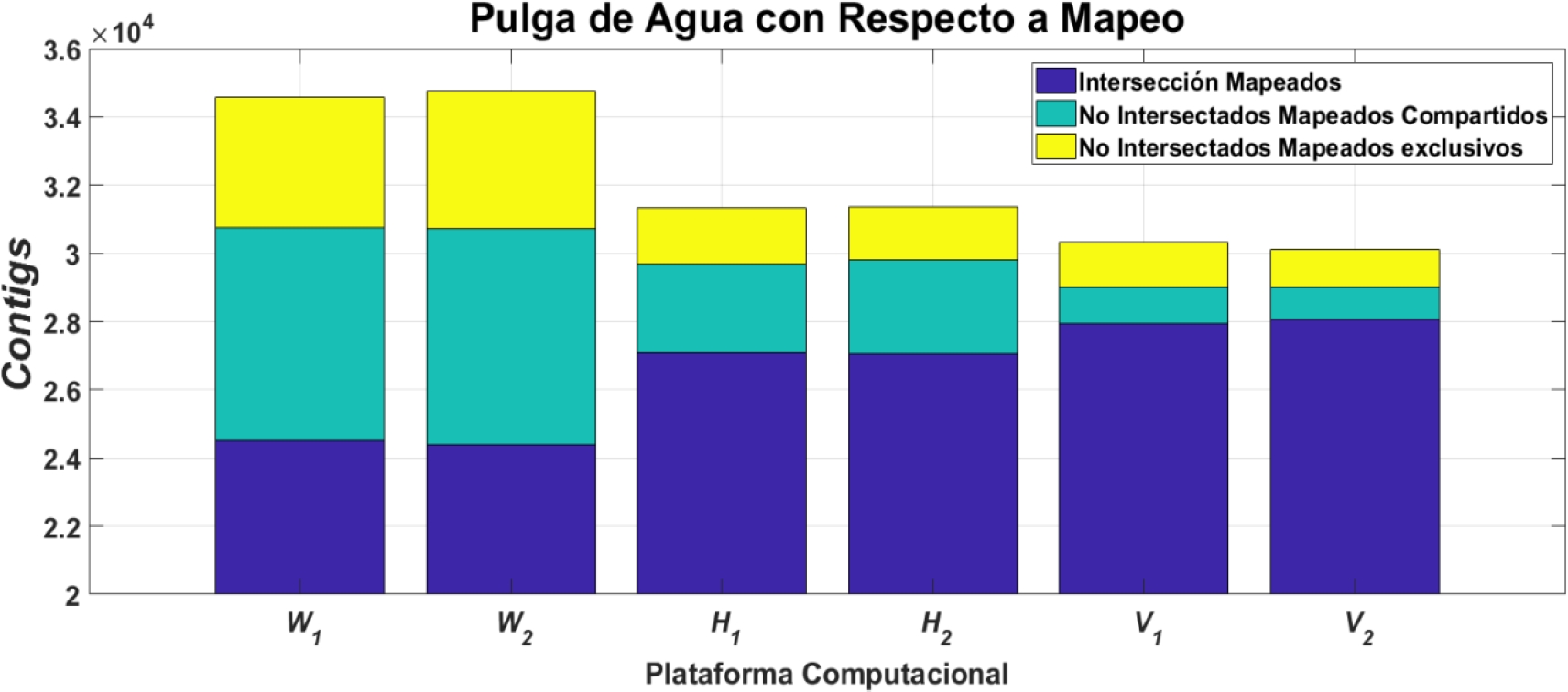
Fig. 6. Comparación de los conjuntos de contigs intersectados mapeados Im
(p,pulga)
al transcriptoma de referencia de la Mosca de la Fruta T
pulga
, contigs no intersectados mapeados exclusivos
Tabla 5 Mapeos de contigs con respecto a las referencias
| Mosca de la Fruta | |||||
|---|---|---|---|---|---|
| Plataforma Computacional |
|
|
|
|
|
| W 1 | 1,186 | 315 | 1,196 | 99.16 | 26.33 |
| W 2 | 1,054 | 312 | 1,070 | 98.50 | 29.15 |
| H 1 | 584 | 160 | 593 | 98.48 | 26.93 |
| H 2 | 583 | 182 | 594 | 98.15 | 30.63 |
| V 1 | 409 | 87 | 418 | 97.85 | 20.81 |
| V 2 | 364 | 86 | 373 | 97.59 | 23.05 |
| Pulga de Agua | |||||
| Plataforma Computacional |
|
|
|
(%) |
(%) |
| W 1 | 10,057 | 3,831 | 13,122 | 76.64 | 29.19 |
| W 2 | 10,369 | 4,017 | 13,682 | 75.79 | 29.35 |
| H 1 | 4,245 | 1,628 | 6,027 | 70.43 | 27.01 |
| H 2 | 4,327 | 1,567 | 6,050 | 71.52 | 25.90 |
| V 1 | 2,385 | 1,323 | 3,918 | 60.87 | 33.76 |
| V 2 | 2,074 | 1,119 | 3,393 | 61.13 | 32.97 |
Máximos entre plataformas W 1, W 2 y en HPC remarcados en gris.
La ganancia máxima de contigs no intersectados mapeados, para Mosca de la Fruta es 1,186/584 = 2.03 veces más que el máximo de una de las plataformas basadas en HPC; asimismo, para Pulga de Agua es 10,057/4,327 = 2.39 veces más.
La ganancia máxima de contigs no intersectados mapeados exclusivos, para Mosca de la Fruta es 315/182 = 1.73 veces más que el máximo porcentaje de las plataformas basadas en HPC; asimismo, para Pulga de Agua es 4,017/1,628 = 2.46 veces más.
4. Discusión
La aproximación inicial de los recursos mínimos computacionales para ensamblaje de novo está dada por los requerimientos básicos del software ensamblador y la cantidad de datos de entrada. El requerimiento mínimo de memoria para ensamblaje de novo de transcriptoma reportado en el ensamblador Trinity es ~ 1GB de memoria por
cada millón de lecturas de entrada [17]. Según esta estimación, para los datos de Mosca de la Fruta (~7.5 millones de lecturas) y Pulga de Agua La memoria en todas las plataformas era mucho mayor que los requerimientos mínimos teóricos para ensamblaje, para los datos en ambos organismos, como se indicó en la Tabla 1. Con base en los requerimientos mínimos teóricos de ensamblaje, la asignación de memoria o memoria disponible por plataforma fue calculada a más del doble o el triple.
En la práctica los requerimientos mínimos reales de memoria para ensamblar excedieron el límite teórico, utilizando más del triple de este; tal como se observa en la Figura 2 (a y b), donde se graficó la utilización de memoria en H 2, y asimismo en la Figura 2 (c y d) para W 2, al ensamblar ambos organismos. El ensamblador tiende a utilizar cuanta memoria esté disponible para realizar sus procesos, independientemente del parámetro de uso de memoria asignado.
Se ha reportado que el primer módulo de Trinity es más extensivo en el uso de memoria [15], lo cual coincide con las gráficas de la Figura 2. Según la asignación de memoria para H 2 el límite de uso debió haber sido muy cercano a 64 GB, pero éste fue excedido por más de 11 GB en ambos organismos (ver Tabla 3).
Caben destacar que los ensambles generados por las plataformas V 1 y H 1, en donde el uso de memoria, asignado en Trinity, estaba limitado a la misma cantidad de GB que en la plataforma W 2, y cuyos conjuntos intersecciones de contigs fueron mayores, tuvieron mayor disponibilidad de memoria, ya que como se demostró con anterioridad, el parámetro de uso de memoria del ensamblador no fue una limitante para la utilización del recurso.
De esta manera, se puede decir que los análisis aquí presentados fueron realizados en plataformas con tres cantidades distintas de memoria 20, 24 y 128 GB, estando representadas las primeras dos en las plataformas de menor memoria (W 1 y W 2, respectivamente) y la última en las de mayor memoria (V 1, V 2, H 1, y H 2), en donde la configuración de memoria por parámetro no fue tomada en cuenta al momento de ejecutar los ensamblajes.
De este modo se empiezan a observar los efectos de las plataformas de cómputo sobre el conjunto de contigs de ensamblajes debido a la disponibilidad de memoria física.
Se observa en la Tabla 2 que las desviaciones estándar indican muy poca variación en la cantidad de contigs construidos, de 2.3 a 6.7 para Mosca de la Fruta y de 15.08 a 39.9 para Pulga de Agua, tendiendo a ser mayores para las plataformas de menor memoria (W 1 y W 2). Si se dejase en este punto la exploración de resultados se encontraría que estas variaciones son muy pequeñas en comparación con la cantidad de contigs obtenidos. Sin embargo, el efecto que los recursos computacionales tuvieron sobre el conjunto de contigs es encontrado en el análisis del contenido de éstos.
En ambos organismos se encontró mayor repetitividad en los ensamblajes procesados por las plataformas con mayor disponibilidad de memoria (V 1, V 2, H 1 y H 2), 97.74% al 98.58%, mientras que en las plataformas con menor memoria (W 1 y W 2) la repetitividad en Mosca de la Fruta fue de 95.51% y 95.97%, respectivamente. Para Pulga de Agua la repetitividad observada en las plataformas con mayor memoria fue del 89.32% al 93.85%, mientras que la repetitividad en las plataformas W 1 y W 2 fue 77.55% y 78.36%. Esto puede ser observado en los conjuntos de contigs intersectados de las Figuras 3 y 4, y en la Tabla 4.
Por otro lado, las plataformas con menor memoria presentaron mayor variabilidad (ver Tabla 4). Para Mosca de la Fruta la mayor variabilidad observada en las plataformas de baja memoria fue de 4.49% (en W 1); en el caso de las plataformas HPC la variabilidad máxima fue de 2.26% (mismo porcentaje en H 1 y W 2). Para Pulga de Agua las plataformas de menor memoria presentaron una variabilidad máxima de 22.45% (en W 2), mientras que para HPC fue del 10.68% (en H 2). Dadas estas variabilidades se observa que las plataformas menores presentan aproximadamente el doble de variabilidad en comparación de las plataformas de mayor memoria, 1.98 y 2.10 veces mayor en Mosca de la Fruta y Pulga de Agua respectivamente, de acuerdo con el cálculo de ganancia por variabilidad entre plataformas de la sección 2.4.1.
Las plataformas con menos memoria produjeron mayor cantidad de combinaciones de contigs. Sin embargo, se necesita determinar si estas secuencias, producto de un algoritmo computacional, tienen correspondencia a un transcrito real.
Como se mencionó con anterioridad, la concordancia entre ensamblajes y los transcriptomas originales se determinó por medio de mapeos a la referencia codificante más próxima de los organismos. De acuerdo con el análisis de resultados realizado, los mapeos de los conjuntos de contigs no intersectados fueron mayores en los conjuntos provenientes de plataformas con poca memoria (Figuras 5 y 6).
También se observa en la Tabla 5, que para la Mosca de la Fruta los mapeos máximos de los conjuntos no intersectados fueron de 99.16% para plataformas con poca memoria (en W 1), mientras que en HPC fue de 98.48% (en H 1). Para la Pulga de Agua los mapeos máximos de los conjuntos no intersectados fue 76.64% (en W 1), mientras que en HPC fue 71.52%(en H 2). Si bien los porcentajes de mapeos son similares para ambos organismos, los contigs no intersectados mapeados en las plataformas de menor memoria son aproximadamente el doble en comparación con los contigs mapeados en las plataformas HPC.
Para Mosca de la fruta la cantidad de contigs mapeados fue 2.03 veces mayor en la estación de trabajo comparando con los mapeos máximos para HPC; en Pulga de Agua mapearon 2.39 veces más contigs en la estación de trabajo comparando con los mapeos máximos en HPC.
Se puede observar también que los porcentajes de mapeos exclusivos provenientes de los conjuntos no intersectados
Cabe mencionar que una referencia constituye la mejor aproximación disponible, en determinada fecha, al transcriptoma de estudio. En este contexto, se puede decir que gran porcentaje de los contigs obtenidos en los ensamblajes de ambos organismos corresponden a transcritos reales.
Algunos contigs no lograron ser mapeados a sus referencias. Sin embargo, no se puede asegurar que dichas secuencias no tengan correspondencia a algún transcrito, ya que las referencias son actualizadas según se profundice en el conocimiento de cierto organismo. Se tiene mayor conocimiento de las secuencias transcriptómicas en Mosca de la Fruta que de los transcritos de Pulga de Agua, ya que Drosophila melanogaster es uno de los organismos modelo más estudiados por la comunidad científica. El hecho de que los mapeos en Pulga de Agua sean menores no significa que las secuencias no mapeadas sean incorrectas, simplemente las secuencias de dichos contigs pueden ser parte de transcritos que aún no son descubiertos por falta de conocimiento sobre la biología molecular del organismo.
Asimismo, se puede mencionar que las ganancias aquí indicadas se presentaron al trabajar en plataformas computacionales con disponibilidad baja de memoria, pero con ~3 veces más que el mínimo teórico reportado[15], bajo las condiciones de viabilidad de plataforma para el procesamiento de conjuntos de lecturas de 7-8 millones de lecturas pareadas y con la unión sin repeticiones de contigs provenientes de 5 ensamblajes.
Dados estos resultados, se evidenció que la variación de un ensamblaje está dada en función de la disponibilidad de memoria del equipo de cómputo; a mayor disponibilidad de memoria menos variación en ensamblaje y a menor disponibilidad de memoria mayor variación en ensamblaje.
Una de las principales ventajas del RNA-Seq (secuenciación NGS de Transcriptoma) es el poder descubrir nuevos transcritos [6]. Tomando esta característica en cuenta y con base en los resultados de este estudio, se sugiere el emplear una estrategia repetitiva de ensamblaje de novo de transcriptoma para el descubrimiento de una mayor cantidad de transcritos.
Dicha estrategia consiste en la obtención de varios ensamblajes bajo condiciones iniciales iguales en plataformas computacionales viables para proceso, pero con baja disponibilidad de memoria; posteriormente, realizar la unión no repetitiva de contigs de múltiples ensamblajes, ya que se logra obtener conjuntos más grandes de contigs de alta calidad, como fue realizado en los contigs obtenidos en las plataformas de menor memoria W 1 y W 2 (ver Figuras 5 y 6).
El efecto de memoria en otras métricas distintas a la cantidad de contigs o su contenido no fueron analizadas en este estudio. Diversas métricas estadísticas buscan dar un indicativo de la calidad de ensamblaje con respecto al transcritptoma original, pero éstas métricas no muestran indicios cuantitativos del desempeño del equipo de cómputo o su influencia en el conjunto de contigs.
Del mismo modo, se hubiese esperado que la repetitividad de las plataformas V 1 y H 1 fuese muy similar dado que tienen la misma cantidad de memoria (128 GB); no obstante, se encontró mayor variación en la plataforma H 1 . Esto pudiese sugerir que, aparte de la memoria RAM disponible para los procesos de ensamblaje, los efectos computacionales en ensamblaje pueden ser influidos en menor proporción por otros recursos, como la memoria cache del procesador (que es menor en el nivel L3 para los procesadores de las plataformas H 1 y H 2). Éste y otros aspectos, como el número de núcleos de procesamiento, necesitan ser estudiados para ampliar el conocimiento de los efectos computacionales en la tarea de ensamblaje.
5. Conclusiones
El ensamblaje de novo de transcriptoma es una etapa clave en estudios exploratorios del contenido de ARN. Es necesario tomar en cuenta los efectos que la plataforma computacional tiene sobre este proceso. Así mismo, el aprovechamiento de las plataformas disponibles para trabajos de investigación prospectiva debe ser potencializado en la etapa de ensamblaje. Un ensamblaje varía en función de la disponibilidad de memoria del equipo de cómputo; menor disponibilidad de memoria origina mayor variación o combinaciones en múltiples ensamblajes. Aún más, los contigs extra originados por dicha variación han mostrado tener correspondencia con transcriptomas de referencia. Tomando ventaja de la viabilidad de datos, disponibilidad de plataforma y recursos computacionales y su influencia en variabilidad del ensamblaje en función de la memoria, se sugiere descubrir mayor cantidad de contigs de calidad al realizar n repeticiones de ensamblajes bajo las mismas condiciones iniciales.

