











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink1. Introducción
Definir las causas por las cuales se genera una enfermedad se ha vuelto una tarea que involucrar diferentes áreas del conocimiento como la medicina, la biología, las matemáticas aplicadas y la informática, generando nuevos estudios basados en grandes cúmulos de información médica que ayudan al descubrimiento de causas relevantes para el comportamiento de una enfermedad. Uno de los métodos más utilizados son los datos de expresión genética obtenidos de la tecnología de microarreglos de ADN [1, 2], ésta tecnología ayuda a comprender la dinámica celular y sus relaciones con estados patológicos [1]. Sin embargo, los datos de expresión genética son difíciles de estudiar, ya que tienen como característica principal una alta dimensión debido a que el número de genes existentes es considerablemente mayor (usualmente miles), en comparación con la cantidad de muestras analizadas (usualmente menos de 100) [3]. En este documento, para abordar la problemática de obtener información relevante de los microarreglos de ADN, se propone un método híbrido de selección y clasificación para realizar una reducción de la dimensión de las bases de datos. Primero se realiza una limpieza del microarreglo utilizando un pre-procesamiento de los datos, eliminando los genes ruidosos y generando una primera reducción de la base genómica.
Después se genera una selección de un subconjunto de genes utilizando una heurística de búsqueda basada en la búsqueda tabú, para evaluar la calidad del subconjunto seleccionado, se utilizan tres métodos de clasificación, con la combinación de estas técnicas, se buscan los genes con información relevante dentro de tres bases de datos obtenidas de microarreglos de ADN de dominio público.
2. Selección y clasificación de datos genómicos utilizando minería de datos
La tecnología de microarreglos de ADN, se utilizan para la adquisición y almacenamiento de datos obtenidos directamente del genoma humano [2]. Permite manipular grandes cantidades de información genética, sin embargo, no toda la información contenida es útil, una base de datos obtenida de la tecnología de microarreglos de ADN, tiene un gran número de características que requieren un largo tiempo de procesamiento para ser analizada. Además, las bases de datos tienen dimensiones altas, miles de los genes son redundantes o contienen ruido [3].
Existen diferentes técnicas basada en minería de datos (como selección y extracción de características), y de aprendizaje máquina [4, 5], que ayudan a obtener información relevante a través de la exploración de los microarreglos de ADN. La selección de características (SC), ayuda a explorar datos de expresión genética que normalmente contienen un número grande de genes, pero un número pequeño de muestras.
La SC se puede ver como el proceso de encontrar un conjunto de genes que determinen mejor las diferencias existentes en una muestra biológica [4]. Además, tiene los objetivos esenciales para reducir el ruido y redundancia de los datos, sirve para mejorar la exactitud de clasificación de una muestra, y los resultados ayudan a biólogos a que se enfoquen en los genes seleccionados para mejorar sus pruebas y validar sus hipótesis biológicas [6]. Para generar una selección de características efectiva se puede ocupar métodos basados en estadística y en aprendizaje máquina [4], los métodos estadísticos seleccionan características basándose en un criterio de discriminación que son relativamente independientes de su clasificación. Los métodos basados en aprendizaje máquina realizan la selección de variables usando como criterio de evaluación la estimación del error basadas en algún clasificador como las redes neuronales o clasificadores bayesianos [7, 8]. La figura 1 muestra un proceso de selección de características, éstas se evalúan en caso que sean características relevantes y se apartan del conjunto original, sino, se vuelve a generar un nuevo subconjunto para ser evaluado, así este proceso se repite un número de veces.

3. Algoritmo híbrido para la exploración de microarreglos de ADN
En este documento se aborda el problema de selección y clasificación efectiva de genes de microarreglos de ADN, utilizando un método híbrido combinando técnicas de selección de características basadas en filtrado de datos como primera etapa de selección. En la segunda etapa de selección y clasificación se utiliza un método híbrido basado en una búsqueda tabú combinada con tres diferentes clasificadores, la figura 2 muestra el método general de selección y clasificación de genes.

3.1. Estandarización de los datos
Las bases de datos obtenidas de la tecnología de microarreglos de ADN no son homogéneas, es decir, la forma en que se encuentra la información original tiene diferentes escalas numéricas y sigue diferentes distribuciones estadísticas.
La estandarización de datos se utiliza para transformar los datos con diferentes distribuciones a una escala igual para todos los datos. En este experimento, los datos son estandarizados utilizando una normalización Min-Max, la cual está definida por [9]:
donde X es la base de datos original. Min(X) y Max(X) son el dato mínimo y el dato máximo existentes en las bases de datos, X’ es la nueva base de datos normalizada.
3.2. Filtrado de datos
Dentro de los microarreglos, existe información poco confiable, que puede sobre entrenar al clasificador y dar resultados erróneos, una de las formas de eliminar esta información es utilizando métodos de limpieza de ruido, al eliminar la información errónea o ruidosa se obtienen muestras bien etiquetadas para clasificar nuevos patrones dentro del microarreglo [10]. En este trabajo se utiliza un pre-procesamiento para eliminar el ruido del microarreglo de la siguiente forma: se genera una primera selección de información para cada base de datos, utilizando una puntuación generada por cinco métodos de filtrado estadístico independientes, esta puntuación sirve como indicador discriminatorio entre los genes para saber cuál de ellos contiene información más relevante [12].
Los cinco filtros que se utilizan en este estudio son: sumas de cuadrados entre los grupos y dentro de los grupos (BSS/WSS), información mutua, relación señal a ruido, prueba de wilcoxon y T-statistic. Estos filtros se utilizan por sus capacidades estadísticas, cada filtro prioriza un gen en particular y los demás filtros priorizan otros genes, se desea hacer un consenso de los genes mejores filtrados con cada uno de los métodos utilizados y con éste consenso trabajar por separado dentro del algoritmo propuesto.
3.2.1. BSS/WSS (BW)
La selección de genes se basa en la razón de las sumas de cuadrados entre los grupos (BSS) y dentro de los grupos (WSS). Para el (gen) j, la razón está dada por [11]:
donde
3.2.2. Información mutua (MI)
Sean A y B dos genes aleatorios con distribuciones de probabilidad diferentes y una distribución de probabilidad conjunta. La información mutua entre ambos genes I(A; B) se define como la entropía relativa entre la probabilidad conjunta y el producto de probabilidades [12]:
donde
3.2.3. Relación señal a ruido (SN)
Identifica los patrones de expresión genética con una diferencia máxima en la expresión media entre dos clases y la variación mínima de expresión dentro de cada clase. En este método, los genes son los primeros clasificados de acuerdo a sus niveles de expresión [13]:
donde
3.2.4. Prueba de Wilcoxon (WT)
Para cada gen j, se necesita el supuesto que las observaciones xij,...,xnj sean independientes. Si rank(xij) denota el rango de xij en la sucesión xij,…,xnj, la prueba estadística para el gen j está dada por [14]:
Para probar la hipótesis se utiliza
H0: mediana
H1: mediana
Bajo H0, Wj tiene una distribución de Wilcoxon con grados de libertad n1 y n2. El valor descriptivo de la prueba (p-value) correspondiente para cada gen j puede ser usado como una medida de relevancia.
3.2.5. T-statistic (TT)
Cada muestra se etiqueta con {1, -1}. Para cada gen
Entonces una puntuación T
donde
3.3. Subselección y clasificación utilizando una búsqueda tabú
Después de generar una primera selección de genes con los métodos de filtro (etapa 1), se hará una nueva selección utilizando una búsqueda tabú combinada con diferentes clasificadores, el método será entrenado con los datos obtenidos por los filtros y se describe a continuación.
3.3.1. Búsqueda tabú
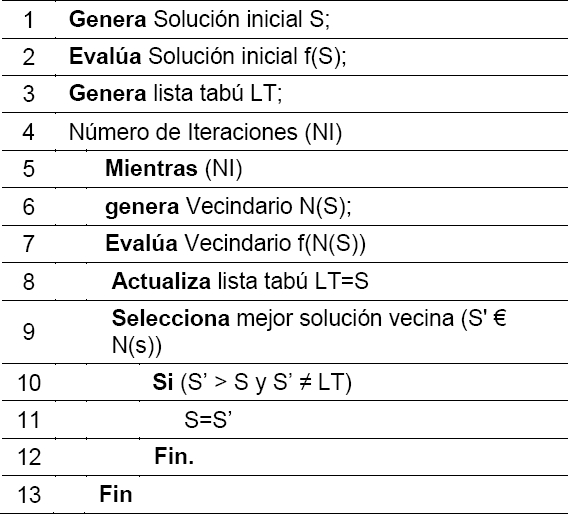
La búsqueda tabú (BT), es una técnica que utiliza una memoria, con el objetivo de guiar un procedimiento de búsqueda local para resolver problemas de optimización combinatoria con un alto grado de dificultad, explorando el espacio de soluciones más allá del óptimo local [16]. Se puede obtener un algoritmo BT básico mediante la utilización de una lista tabú. En cada iteración, la solución actual S es reemplazada por la mejor solución vecina S' que no esté prohibida por la lista tabú. S' Є N(s) de tal manera ∀ s'' Є N(s), f(S") ≤ f(s') y S' donde es el conjunto de soluciones prohibidas por la lista tabú.
Note que el vecino seleccionado S' puede o no puede ser mejor que S. El algoritmo BT se detiene cuando un número fijo de iteraciones se alcanza o cuando todos los movimientos se han convertido en tabú. La principal función de la lista tabú es prevenir que se cicle la búsqueda [17]. EL código simple de una búsqueda tabú se describe a continuación.
El algoritmo muestra un proceso general de una búsqueda tabú, actualizando la lista tabú y reemplazando la solución anterior por la mejor solución encontrada dentro del vecindario.
3.3.2. Máquina de vectores de soporte
El clasificador máquina de vectores de soporte (SVM por Support Vector Machine), es una técnica que opera de acuerdo a un paradigma de aprendizaje supervisado, aprendiendo de una relación funcional entre los atributos (o características), de entrada y salida por medio de apariciones de ejemplos etiquetados, se utiliza para analizar datos y reconocer patrones, para metodologías estadísticas y análisis de regresión, el algoritmo de entrenamiento SVM, construye un modelo que predice si un nuevo ejemplo sigue dentro de una categoría o de otra [13].
Discriminan datos de clases linealmente separables, dibujando un hiperplano óptimo en el espacio del vector de características, de tal manera que maximice el margen de separación entre los ejemplos positivos y negativos [18]. Los clasificadores SVM funcionan de la siguiente forma. Dado un conjunto de muestras m etiquetados S= {(xi, yi) | (xi, yi) ϵ Rn x {±1}, i=1, 2,..., m}, donde xi ϵ Rn, yi ϵ {+ 1} es una etiqueta de la muestra de xi, el hiperplano se define por [19]:
donde
3.3.3. K-vecino más cercano
El clasificador k-vecino más cercano (KNN por k-Nearest Neighbor), es un algoritmo de clasificación que basa su criterio de aprendizaje en la hipótesis de que los miembros de una población suelen compartir propiedades y características con los individuos que los rodean [14], de modo que es posible obtener información descriptiva de un individuo mediante la observación de sus vecinos más cercanos.
La regla de clasificación por KNN se describe a continuación. Sea x
1
, x
2
,…, x
n
una muestra con una función f(x) de densidad desconocida. Se estima f(x) a partir de un elemento central de la muestra
Estas observaciones son los k vecinos más cercanos a
donde
3.3.4. Análisis lineal discriminante (LDA)
El clasificador basado en Análisis Lineal Discriminante (LDA por Lineal Discriminant Analysis), es una técnica de aprendizaje supervisado para clasificar datos. La idea central de LDA es obtener una proyección de los datos en un espacio de menor (o incluso igual), dimensión que los datos entrantes, con el fin de que la separabilidad de las clases sea la mayor posible [21]. LDA se acerca al problema de clasificación mediante la búsqueda de una matriz de transformación que ayude a preservar la mayor parte de la información que se utilice para discriminar entre diferentes clases.
Lo anterior se logra mediante la reestructuración de los datos de alta dimensión proyectándolos en un espacio de pocas dimensiones. Para alcanzar la matriz de transformación óptima, dos matrices S B (dispersión entre las clases) y S W (dispersión dentro de las clases) deben ser calculadas de acuerdo a las siguientes ecuaciones [21]:
donde n k es el número de ejemplos de entrenamiento para la clase k, c k es el conjunto de índices de los ejemplos de entrenamiento pertenecientes a la clase k, x i es el valor de expresión genética del gen i, µ k es la media de la clase k y µ es la media resultante de las dos clases.
Entonces LDA está preparada para clasificar nuevas muestras después de que encontró un valor óptimo para el vector w tal que
3.3.5. Procedimiento general
En nuestro caso el algoritmo es implementado de la siguiente manera:
− La búsqueda tabú se implementa de manera binaria, donde la solución inicial S se genera de forma aleatoria con una distribución uniforme.
− En la función de costo de la búsqueda, se utiliza uno de los tres clasificadores descritos anteriormente, esto servirá para evaluar (f(s)) la calidad de los genes seleccionados por la solución inicial, el resultado obtenido por el clasificador es validado utilizando el método 10-fold cross-validation.
− Se genera el vecindario a partir de la solución inicial S, el cual se evalúa con la función de costo. Se busca dentro del vecindario la mejor solución S’ y se verifica si se encuentra prohibida por la lista tabú, si S’ es tabú, se toma la segunda mejor solución del vecindario, si S’ no es tabú, S’ se toma como la mejor solución del vecindario.
− La solución S’ reemplaza a la solución inicial (S=S’) y se genera un nuevo vecindario a partir de la solución S,
− Este proceso se repite n veces hasta que S’ hasta que se cumplan un número de iteraciones.
− La lista tabú es implementada de la siguiente manera.
− Cada vez que un movimiento mv(i,j) se lleva a cabo, un gen es descartado y un gen es seleccionado, el gen seleccionado es guardado en la lista tabú por las siguientes k iteraciones.
Por consecuencia, este gen no se puede volver a seleccionar durante el proceso.
− El valor de k es el tiempo de permanencia que el gen estará dentro de la lista tabú y varía desde kmin a kmax.
− La lista tabú prohíbe un nuevo gen seleccionado, este gen se puede retirar de la lista tabú (criterio de aspiración), en la siguiente iteración sí el coeficiente de clasificación del nuevo gen seleccionado es muy bajo.
4. Experimentos y resultados
El método propuesto se entrena con diferentes conjuntos de datos genómicos, se logra observar que el método es capaz de seleccionar genes con información relevante.
En esta sección se muestran los resultados obtenidos por el método propuesto y se genera un estudio de comparación de los resultados obtenidos con diferentes literaturas.
4.1. Microarreglos de ADN
Los microarreglos de ADN, son una herramienta que permite realizar diversos análisis genéticos basados en la miniaturización de procesos biológicos, su funcionamiento se basa en la capacidad que tienen las moléculas complementarias de ADN de hibridar entre sí, utilizando pequeñas cantidades de ADN correspondientes a diversos genes cuya expresión se desea medir [1]. Los microarreglos de ADN tiene forma de una matriz de datos donde las filas representa los genes y las columnas representan las muestras.
Cada celda dentro de ésta matriz, es un valor de expresión genética que representa la intensidad del gen correspondiente a cada una de las muestras. Lo anterior se observar en la figura 3 donde x representa el dato genómico, n g (número de gen, filas) los genes dentro de la matriz y n m (número de muestras, columnas) las muestras dentro de la matriz.

En éste trabajo, se utilizan tres microarreglos de ADN descritos en la tabla 1.
4.2. Parámetros
El algoritmo híbrido ha sido implementado en Matlab (Versión 7.11.0). Los parámetros más confiables con los cuales fue entrenada la búsqueda tabú para las tres bases de datos se muestran en la tabla 2.
4.3. Resultados
En el protocolo experimental, los cinco métodos de filtrado de datos funcionan como una etapa de pre-selección generando una reducción significativa de las tres bases genómicas, descartando los genes ruidosos y genes redundantes y obteniendo como resultado los nuevos subconjuntos con información relevante, que se utilizan en la siguiente etapa para entrenar la búsqueda tabú y los clasificadores. En esta nueva etapa se genera una selección de genes dentro los nuevos subconjuntos, eliminando los genes menos informativos y seleccionando los genes que logran entrenar mejor al clasificador. Así se obtiene un subconjunto mínimo de genes con una tasa de desempeño alta.
La figura 4 (leucemia), 5 (cáncer de colon) y 6 (cáncer de pulmón), muestran la comparación de las tasas de clasificación que se han obtenido al entrenar el algoritmo tabú combinado con cada uno de los clasificadores (SVM, LDA, KNN).
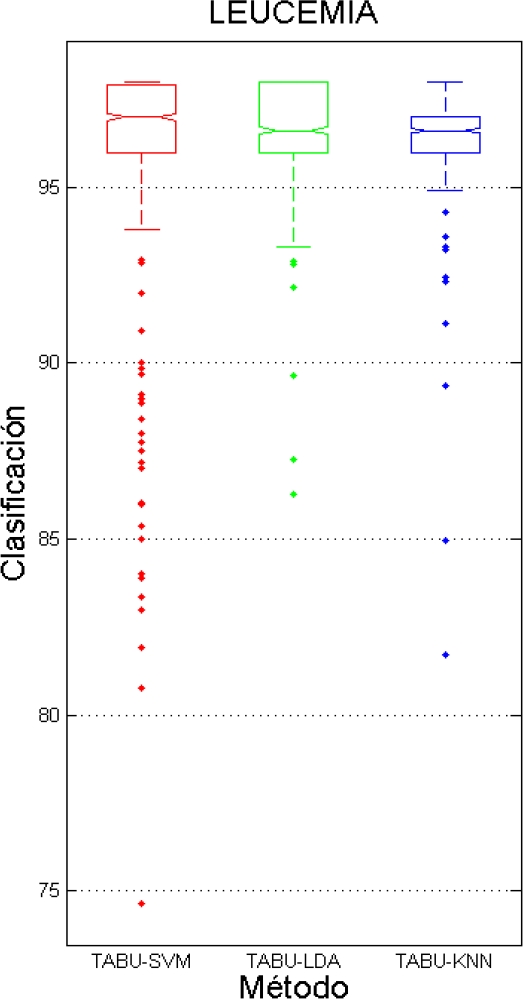

Fig. 5 Tasa de clasificación obtenida por el algoritmo híbrido para la base de datos de cáncer de colon

Fig. 6 Tasa de clasificación obtenida por el algoritmo híbrido para la base de datos de cáncer de pulmón
La tabla 3, muestra la mejor tasa de clasificación obtenida por el método propuesto y la comparación con diferentes métodos reportados en la literatura. La tabla se divide de la siguiente forma: en la primera columna, se muestran los autores con los que se han comparado los resultados obtenidos, el resto de las columnas muestran las tasas de clasificación (%) y el número de genes (Ng), que fueron obtenidos por los métodos propuestos para las tres bases de datos. Los resultados obtenidos se han comparado con los autores mostrados en la tabla 3. Cabe mencionar que algunos autores presentan un modelo basado en algún tipo de metaheurística como un algoritmo genético o se basan en búsquedas locales y otros clasifican basándose en técnicas de aprendizaje máquina.
Tabla 3 Tasas de clasificación obtenidas por el algoritmo híbrido
| AUTORES | Leucemia | Colon | Pulmón |
|---|---|---|---|
| %(Ng) | %(Ng) | %(Ng) | |
| Luo et al. [25] | 71.39 (5) | 80.07(7) | -- |
| Yu et al. [26] | 96.8(10) | 88.6(10) | 94.7(10) |
| Cho et al. [29] | 95.9(25) | 87.7(25) | -- |
| Hernández et al. [17] | 92.52(6) | 87.00(8) | -- |
| Filippone et al. [27] | 94.7(13) | 80.6(21) | -- |
| Li et al. [28] | 95.1(21) | 88.7(16) | -- |
| Bonilla et al. [33] | 99.5(3) | 90.5(3) | 96.0(3) |
| Tan et al. [34] | 91.1 | 95.1 | 93.2 |
| Yue et al.[35] | 83.8(100) | 85.4(100) | -- |
| Pang et al. [31] | 94.1(35) | 83.8(23) | 91.2(34) |
| Li et al. [32] | 97.1(20) | 83.5(20) | -- |
| Zhang et al. [30] | 100(30) | 90.3(30) | 100(30) |
| González [54] | 99.62(3) | 89.19 (5) | 99.89(7) |
| Tabú-SVM | 98.00(4) | 95.90(3) | 97.94(3) |
| Tabú-LDA | 98.00(2) | 93.77(2) | 97.17(3) |
| Tabú-KNN | 98.00(2) | 94.77(3) | 97.72(3) |
En tabla 3 al comparar las tasas de clasificación obtenidas por nuestro método para las tres bases de datos, se aprecia que el método es muy competitivo en relación con algunos de los autores con los que se han comparado.
Por ejemplo, Zhang et al. [30] obtuvo el 100% de clasificación seleccionando un subconjunto de 30 genes para Leucemia, Bonilla-Huerta et al. [33], obtuvo 90.5% de clasificación seleccionando un subconjunto de 3 genes para Cáncer de Colon, Zhang et al. [30], obtuvo el 100% seleccionando un subconjunto de 30 genes para Cáncer de Pulmón.
Los mejores resultados obtenidos para las bases de datos son:
Para Leucemia la tasa de clasificación más alta fue de 98% seleccionando un subconjunto de 2 genes, en Cáncer de Colon la tasa de clasificación más alta fue de 95.90% seleccionando un subconjunto de 3 genes y Cáncer de Pulmón la tasa de clasificación más alta fue 97.94% seleccionando un subconjunto de 3 genes.
En cada una de las tres bases de datos existen genes informativos que entrenan al clasificador de manera eficiente, una forma de verificar si los genes seleccionados pueden ayudar en el diagnóstico de una enfermedad es revisando en la literatura si estos genes han sido reportados por algún autor, de modo que podemos encontrar una interpretación biológica de los genes seleccionados.
La base de datos de leucemia y de cáncer de colon, han sido estudiadas ampliamente, permite encontrar la mayoría de genes relevantes reportados por diferentes autores. Por otra parte, la base de datos de cáncer de pulmón no ha sido estudiado ampliamente, consecuentemente surgen dudas para la comparación de los resultados con los genes reportados. A continuación, se muestran los genes seleccionados y reportados por diferentes autores obteniendo así una interpretación biológica más confiable:
En nuestro trabajo encontramos el gen 4847 (Zyxin) como el más relevante dentro de la base de datos de leucemia, este gen fue seleccionado por los tres métodos utilizados Tabú-(SVM, LDA, KNN). El gen ha sido reportado por [15, 22, 36, 37], [38], indicando que tiene un rol importante dentro de la clasificación de leucemia, debido a su nivel de expresión logra identificar dos tipos de leucemia aguda y así ser clasificado o etiquetado en la clase Leucemia Mieloide Aguda o Leucemia Linfoblastica Aguda. Otros genes encontrados para leucemia son 1882 (CST3 Cystatin C amyloid angiopathy and cerebral hemorrhage) 2020 (FAH Fumarylacetoacetate) y el gen 760 (CYSTATIN A). Los dos primeros fueron encontrados por dos de los tres métodos propuestos Tabú-(SVM, KNN) y Tabú-(SVM, LDA) respectivamente y el último seleccionado por el método (Tabú-SVM). Los tres genes han sido reportados en [15, 29, 37, 38, 39, 40, 41]. Para la base de datos de cáncer de colon cada uno de los métodos ha logrado identificar de dos a tres genes relevantes que ayudan a la clasificación de muestras con tejidos de tumores y muestras de tejidos normales.
Los genes más relevantes que han encontrado son: el gen 245 (Human cysteine-rich protein (CRP) gene, exons 5 and 6) con el método Tabú-(LDA, KNN) reportado en [42, 43, 44] y el gen 765 (Human cysteine-rich protein CRP gene, exons 5 and 6) con el método Tabú-(SVM, KNN) reportado en [37, 42, 45, 46, 47]. Estos dos genes han logrado separar mejor la clase de tejidos de tumores de la clase de tejidos normales, y se pueden utilizar en la identificación células con cáncer de colon. El resto de genes seleccionados para cada método son: para el método Tabú-SVM el gen 249 (Human desmin gene, complete cds), y el gen 897 (3' UTR 2ª 183264 Complement Factor D Precursor (Homo sapiens)). El método tabú-KNN ha seleccionado el gen 267 (Human cysteine-rich protein CRP gene, exons 5 and 6). El método Tabú-LDA también ha seleccionado el gen 493 (Myosin Heavy Chain, Nonmuscle Gallus gallus), estos genes han sido reportados en [33, 37, 46, 48, 49, 50, 51, 52].
En cáncer de pulmón, un total de tres genes relevantes han sido seleccionados por los tres métodos propuestos Tabú-(SVM, LDA, KNN), los genes encontrados son: el gen 3844 (Interferon, alpha-inducible protein clone IFI-6-16), el gen 8537 (Replication protein A1, 70kDa) y el gen 11841 (leucine-rich PPR-motif containing). Estos genes han logrado entrenar mejor los clasificadores, a diferencia de los demás genes utilizados en el estudio. La selección de estos genes se debe a la separación por el clasificador de la información contenida en la base de datos, esto significa que el clasificador ha logrado separar la clase Malignant Pleural Mesothelioma (MPM) de la clase Adenocarcinoma (ADCA). Estos genes han sido reportados en [33, 36, 53].
5. Conclusiones
En este trabajo, se presentó un método híbrido basado en una búsqueda local y técnicas de minería de datos, implementado en la selección y clasificación de un conjunto de genes importantes explorando dentro de tres bases de datos de dominio público (Leucemia, Cáncer de pulmón, y Cáncer de Colon). El método propuesto tiene una etapa de pre-selección de genes mediante la utilización de cinco técnicas de filtrado de datos, estos filtros utilizan una puntuación o categoría que sirve para discriminar los genes contenidos en la base de datos, así se eliminan los genes no relevantes (ruidosos o redundantes) y son seleccionados los genes con información pertinente.
Con lo anterior se ha generado una primera reducción efectiva de la dimensión de las bases de datos. Para realizar la selección dentro de los subconjuntos obtenidos por las técnicas de filtrado, se ha creado un algoritmo híbrido basado en una búsqueda tabú como método de selección de genes combinada con tres técnicas de clasificación (SVM, LDA, KNN). Utilizando las propiedades de memoria de la búsqueda tabú, se ha logrado crear un algoritmo guiado que recuerda los genes que han sido utilizados en un proceso (iteración) anterior. Basándose en la tasa de clasificación del gen recordado permite que el algoritmo prohíba genes que han sido utilizados durante su ejecución y trabaje con nuevos genes logrando explorar a profundidad la base de datos y consecuentemente obtener la mejor tasa de clasificación.
Cada técnica utilizada en éste trabajo ha seleccionado un subconjunto de genes con una tasa de clasificación alta. Para saber la relevancia que tiene cada gen seleccionado, se utiliza la frecuencia de selección del gen por cada método propuesto, de esta manera, se logra observar que un gen en particular al ser seleccionado logra entrenar el clasificador obteniendo una tasa de clasificación aceptable.
El método propuesto determina una tasa de clasificación alta, obtenida con un subconjunto de genes pequeño para las tres bases de datos. Para evaluar la eficiencia del método, se genera un estudio de comparación de los resultados obtenidos con otros métodos reportados en la literatura, esto permite verificar si el método es competitivo.
Se observa que en algunos casos se ha logrado superar las tasas de clasificación y se han obtenido un subconjunto de genes pequeño en comparación de los métodos reportados. Además de las tasas de clasificación, se desea conocer si los genes han sido reportados en la literatura, esto permite tener una mejor interpretación biológica de los genes que ha seleccionado el algoritmo.
También se ha minimizado el número de genes a utilizar y en algunos casos igualado la exactitud de la clasificación utilizando la búsqueda Tabú con uno de los tres clasificadores (SVM, KNN, LDA) dentro del proceso de minería de datos. En trabajos futuros, se pretende probar y compara otros algoritmos de selección de características, también utilizar otros métodos de clasificación, la meta es minimizar el número de genes a utilizar y maximizar la tasa clasificación.

