










Services on Demand
Journal

Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
 Cited by SciELO
Cited by SciELO Related links
 Similars in
SciELO
Similars in
SciELO Share

 Permalink
PermalinkRevista de la Sociedad Química de México
Print version ISSN 0583-7693
Rev. Soc. Quím. Méx vol.48 n.4 Ciudad de México Oct./Dec. 2004
Investigación
Preparation of N-Acylbenzotriazole Derivatives of Dicarboxylic Acids
Alan R. Katritzky,* Nabin K. Meher, Chunming Cai, and Sandeep K. Singh
Center for Heterocyclic Compounds, Department of Chemistry, University of Florida, Gainesville, FL 32611-7200, USA.
Recibido el 29 de octubre del 2004.
Aceptado el 12 de noviembre del 2004.
Abstract
N-Acylbenzotriazole derivatives of dicarboxylic acids have been prepared by convenient methods.
Key words: N-acylbenzotriazole, dicarboxylic acids, synthesis.
Resumen
Se describe la preparación de N-acilbenzotriazol derivado de ácidos carboxílicos.
Palabras clave: N-acilbenzotriazol, ácidos dicarboxílicos, síntesis.
Dedicated to the memory of Dr Raymundo Cruz Almanza†
Introduction
N-Acylbenzotriazoles are important: (i) as C-acylation reagents for the synthesis of 1,3- [1a] and 1,2-diketones, [1b] for the conversion of imines into enaminones, [1c] and for the regiospecific acylation of heterocycles; [1d,e] (ii) as neutral N-acylation reagents including formylation [2a] and trifluoroacylation; [2b] for the preparation of amides [3a-c] and peptides; [3d] (iii) as O-acylation reagents in additions to aldehydes to give esters; [4] (iv) in the preparation of benzoxazoles by flash vacuum pyrolysis (FVP); [5] and (v) in the syntheses of oxazolines and thiazolines under microwave irradiation (MW) [6] (Scheme 1).
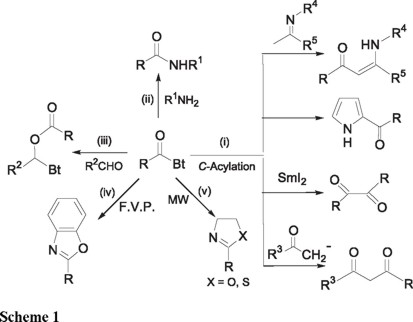
Use of N-acylbenzotriazoles avoids racemization, [3b,d] assures regiospecificity [1d,e], and generally provides products in high yields. Unlike acid chlorides, N-acylbenzotriazoles are stable crystalline compounds that can be stored at room temperature without decomposition. Literature reports on the applications of N-acylbenzotriazoles in the last decade show a wide variety of organic transformations in which they have been used advantageously in place of acid chlorides. They are also the reagents of choice when the corresponding acid chlorides are unstable or difficult to isolate, for example RCOCl, with R = 4-diethylaminophenyl, 2-pyridyl, 2-indolyl or 2-pyrrolyl etc.
We reported a mild one-pot procedure for efficient conversion of carboxylic acids into the corresponding N-acylbenzotriazoles [7] that has several advantages over the previous methods [3a,8]. We recently found that these procedures require further modifications for the preparation of N-acylbenzotriazoles of important dicarboxylic acids which have low solubility; the present work involves the preparation of this class of derivatives.
Results and Discussion
Preparation of N-Acylbenzotriazole Derivatives. Reaction of 1 equivalent of glutaric acid, 8 equivalents of benzotriazole and 2 equivalents of SOCl2 in CH2Cl2 for 2 h according to the previous procedure [7] gave a very low yield of the corresponding N-acylbenzotriazole derivative 1a. Increasing the reaction time to 48 h gave 1a in 60% yield but a substantial amount of the diacid remained undissolved. Changing the solvent to THF gave 1a in the same yield in 48 h but the diacid appeared to be more soluble in THF as compared to CH2Cl2. Interestingly, using THF as the solvent, the reaction of diglycolic acid, benzotriazole and SOCl2 gave the corresponding N-acylbenzotriazole derivative 1b in 98% yield in 24 h. Similar reactions with thiodiglycolic acid and trans-1,4-cyclohexanedicarboxylic acid gave N-acylbenzotriazoles 1c and 1d in 87% and 16% yields, respectively. Using this procedure, reactions of methylmalonic acid, phenylmalonic acid, diphenic acid and 3,3'-dithiodipropionic acid each gave the corresponding N-acylbenzotriazoles 1e (80%), 1f (82%), 1g (94%) and 1h (98%), respectively. However, derivative 1i from 3,3-dimethylglutaric acid was obtained in a low yield (40%), and the reaction of fumaric acid resulted in mono-derivatization to give (2E)-4-1H-1,2,3-benzotriazol-1-yl)-4-oxobut-2-enoic acid (1j) in 65% yield (Table 1).
The above modification in the one pot procedure did not give satisfactory yields of N-acylbenzotriazole derivatives in the case of benzenedicarboxylic acids; these derivatives were prepared by the reaction of 1-(methylsulfonyl)benzotriazole [3a] with a salt of the carboxylic acid. Thus, reaction of 1 equivalent of 1,4-benzenedicarboxylic acid with 2 equivalents of 1-(methylsulfonyl)benzotriazole in presence of 2 equivalents of triethylamine in refluxing THF for 24 h gave the corresponding N-acylbenzotriazole derivative 1k in 80% yield. Similarly, reaction of 1,3-benzenedicarboxylic acid gave 1l in 41% yield. The N-acylbenzotriazole derivative of 1,2-benzenedicarboxylic acid, 1m was prepared by the reaction of commercially available phthaloyl chloride with benzotriazole at 25 °C in 2 h. (Table 1). All of the N-acylbenzotriazole derivatives prepared except 1b and 1k-l are novel compounds and have been fully characterized by 1H and 13C NMR spectroscopy and elemental analysis.
Experimental
Melting points are uncorrected. 1H and 13C NMR spectra were recorded at 300 and 75 MHz, respectively, with tetramethylsilane (TMS) as an internal standard. Solvents were distilled by standard methods. Reagents obtained commercially were used without further purification.
General procedure:
Method A (for 1a-1j): To a solution of BtH (9.6 g, 80 mmol) in THF (100 ml), SOCl2 (1.5 mL, 20 mmol) was added drop-wise with stirring at room temperature. After 30 min, a solution of dicarboxylic acid (10 mmol) in THF (50 mL) was added. After 24-48 h (Table 1), the solid was filtered and washed with THF (50 mL). The solvent was removed under vacuum from the combined filtrate. To the residue, CHCl3 (150 mL) was added; the mixture was washed with water (30 ml) and saturated Na2CO3 (3 × 30 mL). The organic layer was dried over anhydrous Na2SO4, then it was filtered and the solvent was evaporated under vacuum to obtain a solid, which was recrystallized from an appropriate solvent or solvent mixture to obtain the pure product.
1,5-Di(1H-1,2,3-benzotriazol-1-yl)-1,5-pentanedione (1a). Colorless prisms; mp 188-189 °C; 1H NMR (CDCl3, 300 MHz) δ 2.55 (quintet, J = 7.2 Hz, 2H), 3.70 (t, J = 7.2 Hz, 4H), 7.52 (dd, J = 8.1, 6.9 Hz, 2H), 7.67 (dd, J = 8.1 Hz, 6.9 Hz, 2H), 8.13 (d, J = 8.4 Hz, 2H), 8.29 (d, J = 8.1 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 18.8, 34.7, 114.6, 120.4, 126.5, 130.7, 131.3, 146.4, 171.8. Anal. Calcd for C17H14N6O2 requires C, 61.07; H, 4.22; N, 25.14. Found: C, 60.98; H, 4.05; N, 25.22 %.
1-(1H-1,2,3-Benzotriazol-1-yl)-2-[2-(1H-1,2,3-benzotriazol-1-yl)-2-oxoethoxy]-1-ethanone (1b).9 Colorless plates; mp 142-144 °C; 1H NMR (CDCl3, 300 MHz) δ 5.56 (s, 4H), 7.54 (dd, J = 8.1, 6.9 Hz, 2H), 7.70 (dd, J = 8.1, 6.9 Hz, 2H), 8.13 (d, J = 8.1 Hz, 2H), 8.29 (d, J = 8.1 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 70.3, 114.2, 120.6, 126.8, 131.0, 131.1, 146.0, 168.2. Anal. Calcd for C16H12N6O3 requires C, 57.14; H, 3.60; N, 24.99. Found: C, 57.45; H, 3.49; N, 25.00 %.
1-(1H-1,2,3-Benzotriazol-1-yl)-2-{[2-(1H-1,2,3-benzotriazol-1-yl)-2-oxoethyl]sulfanyl}-1-ethanone (1c). Pale yellow prisms; mp 150-160 °C; 1H NMR (CDCl3, 300 MHz) δ 4.58 (s, 4H), 7.54 (dd, J = 8.1, 6.9 Hz, 2H), 7.68 (dd, J = 8.1, 6.9 Hz, 2H), 8.13 (d, J = 8.1 Hz, 2H), 8.23 (d, J = 8.1 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 34.8, 114.5, 120.6, 126.7, 131.0, 131.3, 146.5, 167.9. Anal. Calcd for C16H12N6O2S requires C, 54.54; H, 3.43; N, 23.85. Found: C, 54.75; H, 3.30; N, 23.78 %.
trans-1H-1,2,3-Benzotriazol-1-yl[4-(1H-1,2,3-benzotriazol-1-ylcarbonyl)cyclohexyl]methanone (1d). White flakes; mp 247 °C; 1H NMR (CDCl3, 300 MHz) δ 1.94-2.08 (m, 4H), 2.35-2.41 (m, 4H), 4.05 (m, 2H), 7.53 (ddd, J = 8.1, 6.9, 0.8 Hz, 2H), 7.69 (ddd, J = 8.4, 6.9, 0.8 Hz, 2H), 8.15 (d, J = 8.1 Hz, 2H), 8.33 (d, J = 8.4 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 28.3, 42.8, 114.8, 120.4, 126.5, 130.7, 131.4, 146.4, 174.9. Anal. Calcd for C20H18N6O2 requires C, 64.16; H, 4.85; N, 22.45. Found: C, 64.41; H, 4.75; N, 22.12 %.
1,3-Di(1H-1,2,3-benzotriazol-1-yl)-2-methyl-1,3-propanedione (1e). Colorless needles; mp 158.0-160.0 °C; 1H NMR (CDCl3, 300 MHz) δ 1.98 (d, J = 7.2 Hz, 3H), 6.25 (q, J = 7.2 Hz, 1H), 7.54 (dd, J = 8.1, 6.9 Hz, 2H), 7.70 (dd, J = 8.4, 6.9 Hz, 2H), 8.13 (d, J = 8.4 Hz, 2H), 8.28 (d, J = 8.1 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 14.0, 48.1, 114.5, 120.6, 126.8, 131.1, 131.3, 146.6, 168.7. Anal. Calcd for C16H12N6O2 requires C, 60.00; H, 3.78; N, 26.44. Found: C, 59.95; H, 3.54; N, 26.28 %.
1,3-Di(1H-1,2,3-benzotriazol-1-yl)-2-phenyl-1,3-propanedione (1f). Colorless plates; mp 193-195 °C; 1H NMR (CDCl3, 300 MHz) δ 7.40-7.47 (m, 3H), 7.52 (dd, J = 8.1, 6.9 Hz, 2H), 7.63 (s, 1H), 7.67 (dd, J = 8.4, 6.9 Hz, 2H), 7.75 (d, J = 7.2 Hz, 2H), 8.11 (d, J = 8.1 Hz, 2H), 8.26 (d, J = 8.4 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 58.8, 114.5, 120.7, 126.9, 129.3, 129.5, 130.0, 130.7, 131.1, 131.4, 146.6, 166.3. Anal. Calcd for C21H14N6O2 requires C, 65.96; H, 3.69; N, 21.98. Found: C, 65.99; H, 3.57; N, 22.02 %.
1H-1,2,3-Benzotriazol-1-yl[2'-(1H-1,2,3-benzotriazol-1-ylcarbonyl)[1,1'-biphenyl]-2-yl]methanone (1g). Colorless prisms; mp 223-225 °C; 1H NMR (CDCl3, 300 MHz) δ 7.41-7.50 (m, 4H), 7.54-7.60 (m, 4H), 7.63-7.67 (m, 4H), 7.99 (d, J = 7.8 Hz, 2H), 8.16 (d, J = 8.1 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 114.9, 120.0, 126.4, 127.6, 130.5, 130.6, 131.3, 131.6, 131.9, 132.2, 140.6, 145.9, 167.2. Anal. Calcd for C26H16N6O2 requires C, 70.26; H, 3.63; N, 18.91. Found: C, 70.18; H, 3.50; N, 18.93 %.
1-(1H-1,2,3-Benzotriazol-1-yl)-3-{[3-(1H-1,2,3-benzotriazol-1-yl)-3-oxopropyl]disulfanyl}-1-propanone (1h). Colorless prisms; mp 104-105 °C; 1H NMR (CDCl3, 300 MHz) δ 3.27 (t, J = 6.9 Hz, 4H), 3.90 (t, J = 6.9 Hz, 4H), 7.50 (dd, J = 8.1, 6.9 Hz, 2H), 7.64 (dd, J = 8.1, 7.2 Hz, 2H), 8.10 (d, J = 8.4 Hz, 2H), 8.25 (d, J = 8.4 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 32.2, 35.6, 114.5, 120.4, 126.4, 130.7, 131.1, 146.3, 170.6. Anal. Calcd for C18H16N6O2S2 requires C, 52.41; H, 3.91; N, 20.37. Found: C, 52.46; H, 3.78; N, 20.28 %.
1,5-Di(1H-1,2,3-benzotriazol-1-yl)-3,3-dimethyl-1,5-pentanedione (1i). Colorless prisms; mp 98-100 °C; 1H NMR (CDCl3, 300 MHz) δ 1.42 (s, 6H), 3.84 (s, 4H), 7.50 (dd, J = 8.4, 6.9 Hz, 2H), 7.64 (dd, J = 7.8, 7.5 Hz, 2H), 8.11 (d, J = 8.4 Hz, 2H), 8.27 (d, J = 8.4 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 28.6, 34.1, 44.4, 114.6, 120.2, 126.2, 130.4, 131.1, 146.3, 171.0. Anal. Calcd for C19H18N6O2 requires C, 62.97; H, 5.01; N, 23.19. Found: C, 63.01; H, 5.03; N, 23.37 %.
(E)-4-(1H-1,2,3-Benzotriazol-1-yl)-4-oxo-2-butenoic acid (1j). Colorless needles; mp 183 °C; 1H NMR (DMSO-d6, 300 MHz) δ 7.05 (d, J = 15.6 Hz, 1H), 7.63 (dd, J = 8.2, 6.9 Hz, 1H), 7.80 (dd, J = 8.2, 6.9 Hz, 1H), 8.12 (d, J = 15.6 Hz, 1H), 8.25 (d, J = 8.2 Hz, 1H), 8.26 (d, J = 8.2 Hz, 1H); 13C NMR (DMSO-d6, 75 MHz) δ 114.2, 120.3, 127.0, 130.6, 131.1, 131.2, 136.4, 145.7, 162.3, 165.6. Anal. Calcd for C10H7N3O3 requires C, 55.30; H, 3.25; N, 19.35. Found: C, 55.30; H, 3.11; N, 19.38 %.
Method B (for 1k-1l):
A mixture of 1-(methylsulfonyl)-1H-benzotriazole (3.94 g, 20 mmol), dicarboxylic acid (10 mmol) and triethylamine (3.0 g, 30 mmol) in THF (40 ml) was heated to reflux for 24 h. Then the solvent was evaporated under vacuum and the residue was dissolved in chloroform. The organic layer was washed with water and brine and dried over anhydrous MgSO4. Filtration and evaporation of the solvent under vacuum gave the crude product, which was purified by column chromatography (silica gel) to obtain the pure product.
1H-1,2,3-Benzotriazol-1-yl[4-(1H-1,2,3-benzotriazol-1-ylcarbonyl)phenyl]methanone (1k). White micro crystals; mp 232-234 °C [lit. [10] mp 238-242 °C]; 1H NMR (CDCl3, 300 MHz) δ 7.60 (dd, J = 8.1, 6.9 Hz, 2H), 7.77 (dd, J = 8.2, 6.9 Hz, 2H), 8.21 (d, J = 8.2 Hz, 2H), 8.42 (s, 4H), 8.45 (d, J = 8.4 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 114.8, 120.4, 126.7, 130.8, 131.4, 132.1, 135.6, 145.9, 165.8.
1H-1,2,3-Benzotriazol-1-yl[3-(1H-1,2,3-benzotriazol-1-ylcarbonyl)phenyl]methanone (1l). White micro crystals; mp189-191 °C [lit. [10] mp 199-201 °C]; 1H NMR (CDCl3, 300 MHz) δ 7.59 (dd, J = 8.2, 6.9 Hz, 2H), 7.76 (dd, J = 8.3, 6.9 Hz, 2H), 7.84 (t, J = 7.8 Hz, 1H), 8.19 (d, J = 8.2 Hz, 2H), 8.44 (d, J = 8.3 Hz, 2H), 8.56 (dd, J = 7.8, 1.7 Hz, 2H), 9.07 (br s, 1H); 13C NMR (CDCl3, 75 MHz) δ 114.8, 120.3, 126.6, 128.7, 130.7, 132.0, 132.2, 135.0, 136.2, 145.8, 165.5.
Method C (for 1m):
To a solution of benzotriazole (4.88 g, 41 mmol) in dry THF (20 mL), phthaloyl chloride (2.03 g, 10 mmol) was added dropwise with stirring. After 2 h, the precipitate was filtered. The solvent was evaporated under vacuum to obtain the crude product, which was recrystallized from chloroform/hexane to obtain 1H-1,2,3-benzotriazol-1-yl[2-(1H-1,2,3-benzotriazol-1-ylcarbonyl)phenyl]methanone (2.17 g, 59%).
1H-1,2,3-Benzotriazol-1-yl[2-(1H-1,2,3-benzotriazol-1-ylcarbonyl)phenyl]methanone (1m). [11] White micro crystals; mp 167-169 °C; 1H NMR (CDCl3, 300 MHz) δ 7.49 (ddd, J = 8.4, 6.9, 0.9 Hz, 2H), 7.63 (ddd, J = 8.2, 6.9, 0.9 Hz, 2H), 7.82-7.88 (m, 2H), 8.07 (d, J = 8.2 Hz, 2H), 8.12-8.17 (m, 2H), 8.22 (d, J = 8.2 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 114.5, 120.3, 126.5, 130.6, 131.5, 131.6, 132.1, 133.9, 145.9, 166.4.
References
1. (a) Katritzky, A. R.; Pastor, A. J. Org. Chem. 2000, 65, 3679-3682. [ Links ] (b) Wang, X.; Zhang, Y. Tetrahedron Lett. 2002, 43, 5431-5433. [ Links ] (c) Katritzky, A. R.; Fang, Y.; Donkor, A.; Xu, J. Synthesis 2000, 2029-2032. [ Links ] (d) Katritzky, A. R; Suzuki, K.; Singh, S. K.; He, H.-Y. J. Org. Chem. 2003, 68, 5720-5723. [ Links ] (e) Katritzky, A. R; Suzuki, K.; Singh, S. K. Croat. Chim. Acta 2004, 77, 175-178. [ Links ]
2. (a) Katritzky, A. R.; Chang, H.-X.; Yang, B. Synthesis 1995, 503-505. [ Links ] (b) Katritzky, A. R.; Yang, B.; Semenzin, D. J. Org. Chem. 1997, 62, 726-728. [ Links ]
3. (a) Katritzky, A. R.; He, H.-Y.; Suzuki, K. J. Org. Chem. 2000, 65, 8210-8213. [ Links ] (b) Katritzky, A. R.; Wang, M.; Yang, H.; Zhang, S.; Akhmedov, N. G. Arkivoc 2002, viii, 134-142. [ Links ] (c) Katritzky, A. R.; Yang, H.; Zhang, S.; Wang, M. Arkivoc 2002, xi, 39-44. [ Links ] (d) Katritzky, A. R; Suzuki, K.; Singh, S. K. Synthesis 2004, in press.
4. Katritzky, A. R.; Pastor, A.; Voronkov, M. V. J. Heterocycl. Chem. 1999, 36, 777-781. [ Links ]
5. Baradarani, M. M.; Khalafy, J.; Prager, R. H. Aust. J. Chem. 1999, 52, 775-780. [ Links ]
6. Katritzky, A. R; Cai, C.; Suzuki, K.; Singh, S. K. J. Org. Chem. 2004, 69, 811-814. [ Links ]
7. Katritzky, A. R.; Zhang, Y.; Singh, S. K. Synthesis 2003, 2795-2798. [ Links ]
8. Katritzky, A. R.; Shobana, N.; Juliusz, P.; Pernak, J.; Afridi, A. S.; Fan, W.-Q. Tetrahedron 1992, 48, 7817-7822. [ Links ]
9. Guan, J.-Q.; Shi, M.-L.; Zou, Z.-C.; Ren, B.-Q.; Zhu, J.-F. Youji Huaxue 1994, 14, 507-510; [ Links ] Chem. Abstr. 1995, 122, 81242. [ Links ]
10. Lu, C. X.; Li, Q.; Pan, J. J. Polymer Sc., Polymer Chem. Ed. 1985, 23, 3031-3044. [ Links ]
11 Smith, G.; Bottle, S. E.; Reid, D. A.; Schweinsberg, D. P.; Bott, R. C. Acta Cryst. 2001, E57, 695-696. [ Links ]
Nota
† Post doctoral fellow in the Katritzky group from 1977-1978.

