










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de la Sociedad Química de México
Print version ISSN 0583-7693
Rev. Soc. Quím. Méx vol.48 n.1 Ciudad de México Jan./Mar. 2004
Revisión
Plegamiento de las proteínas: Un problema interdisciplinario
Luis Olivares-Quiroz1 and Leopoldo García-Colín Scherer2
Departamento de Física. Universidad Autónoma Metropolitana-Iztapalapa. Av. Michoacán y Purísima S/N. México D.F. 09340 México. 1Correo-E: luis@abaco.izt.uam.mx
2Correo-E: lgcs@xanum.uam.mx
Recibido el 16 de enero del 2004.
Aceptado el 8 de enero del 2004.
Resumen
La predicción teórica realizada por L. Pauling en 1951 de la existencia de un estado termodinámicamente estable con estructura helicoidal para ciertos tipos de proteínas, establece por primera vez un puente entre los campos de la Biología Molecular y la Física. La confirmación experimental de este hecho, observada primero por Watson y Crick para la molécula del ADN en 1953 y posteriormente por Kendrew para la molécula de mioglobina en 1958, establece simultáneamente la relación existente entre función y estructura. Un ejemplo de esta relación es el hoy conocido problema del plegamiento de una proteína (protein folding), el cual se refiere a la elucidación de los mecanismos a través de los cuales una proteína adquiere una configuración tridimensional unívoca y termodinámicamente estable. La existencia del estado plegado es imprescindible para que pueda ejercer sus funciones biológicas. En este trabajo presentamos una revisión de las ideas principales que han ubicado al problema del plegamiento de las proteínas como uno de los retos para la Física, la Química y la Biología del siglo XXI.
Palabras clave: Plegamiento de proteínas, paisaje energético, vidrios de espín, heteropolímeros, estado nativo, interacción hidrofóbica, termodinámica de proteínas.
Abstract
L Pauling's theoretical prediction (1951) on the existence of a thermodynamically stable helicoidal configuration for certain biological molecules, proposes for the first time a bridge between Molecular Biology and Physics. The experimental confirmation of this fact came out from both Watson and Crick in 1953 for DNA molecule and Kendrew et al. in 1958 for the myoglobine molecule. Simultaneously, these remarkable experiments provided solid evidence for the form-function paradigm. An outstanding example of this paradigm is the so called protein folding mechanism. In this process, a one-dimensional chain of aminoacids is transformed into three-dimensional structure with biological activity. In this work we review some of physical approaches developed over the past decades to shed some light into one of the greatest challenges faced by Physics, Chemistry and Biology along the last century.
Key words: Protein folding, energy landscape, spin glasses, heteropolymer freezing transition, hydrofobic interaction, native state, protein thermodynamics.
Introducción
En 1951, L. Pauling [1] realiza una predicción teórica que vendrá a redefinir la relación existente entre las diversas áreas de la Biología Molecular y la Física. Extendiendo los argumentos de minimización energética de sistemas cristalinos al caso de macromoléculas biológicas, Pauling propone la existencia de un estado termodinámicamente estable con estructura helicoidal para ciertas proteínas. La confirmación experimental de este hecho, llevada a cabo por Watson y Crick [2] en 1953 para la molécula del ADN y por Kendrew et al [3] en 1958 para la molécula de la mioglobina ponen también de manifiesto por primera vez la relación existente entre función y estructura.
En la actualidad se sabe que las proteínas se sintetizan dentro de la célula mediante una acción conjunta entre el ADN y ARN generando una cadena lineal de aminoácidos única para cada proteína, cuya estructura asemeja a un hebra unidimensional suspendida dentro de un fluido. Esta configuración, conocida en la literatura como estructura primaria (unfolded state), no es única y las posibles conformaciones que puede adquirir son similares a las diversas conformaciones posibles para una cadena homopolimérica suspendida dentro de un fluido, como ha sido mostrado en los trabajos clásicos de Flory [4],[5], y De Gennes [6]
Una vez definida la secuencia específica de aminoácidos que integra la proteína, la estructura primaria transita por una serie de estados en los cuales su configuración espacial se modifica, hasta alcanzar un estado termodinámicamente estable con una estructura tridimensional particular, la cual depende de la secuencia de aminoácidos inicial y por consecuencia es distinta para cada proteína. Esta estructura, conocida como estado nativo (folded state), define los sitios activos de la macromolécula1 y es la única relevante desde el punto de vista biológico. Solamente en el estado terciario la proteína es capaz de realizar las diversas funciones bioquímicas para las cuales fue diseñada. En una serie de experimentos cruciales realizados en 1961 por C. Anfinsen [7], se pone de manifiesto por primera vez que el proceso mediante el cual la cadena lineal de aminoácidos adquiere su estructura terciaria es un proceso reversible independiente del medio celular, es decir, puede ser reproducido en el laboratorio. En forma específica, Anfinsen demuestra que los enlaces disulfuros de la ribonucleasa A (enlaces claves en la conformación de su estructura terciaria) se forman de manera espontánea mediante una reacción de oxidación en presencia de aire. Este trabajo ubica al problema del plegamiento de la proteínas dentro del campo de estudio de los procesos físicos y da inicio a una serie de investigaciones que pretenden establecer, bajo bases físicoquímicas, la naturaleza de los mecanismos involucrados.
El estudio del plegamiento de las proteínas no sólo es relevante desde el punto de vista físico y/o bioquímico per se, sino que posee profundas repercusiones en otros campos. Estudios recientes han demostrado que para ciertos padecimientos como la fibrosis quística y el enfisema pulmonar familiar, existe una conexión clara entre el plegamiento incorrecto de una proteína y una disfunción celular específica [8]. Asociado también a un plegamiento incorrecto, se encuentra toda una categoría de disfunciones celulares conocidas con el nombre genérico de amiloidosis, en las que los plegamientos incorrectos conducen a la formación irreversible de agregados fibrilares insolubles llamados amiloides, los cuales interfieren con el desarrollo de las diversas funciones celulares. Dentro de este grupo de patologías se encuentran las diversas variantes de la enfermedad de Alzheimer y Creutz-Jakob [9], [10], entre otras. Quizás una de las características principales de las patologías amiloides es la transformación irreversible de la estructura terciaria de la proteína a una conformación integrada primordialmente por hebras β. En el caso de las enfermedades infecciosas, normalmente se piensa en el agente infeccioso como un complejo macromolecular compuesto de ácidos nucleicos y proteínas, como es el caso de los virus. Sin embargo, análisis recientes plantean la posibilidad de que ciertas proteínas puedan actuar como agentes infecciosos, en el sentido de que una proteína plegada incorrectamente pueda propagar a sus vecinas la malformación. Para designar a este tipo de proteínas infecciosas, se ha acuñado el término prión, con el cual se denomina a una proteína que ha perdido la habilidad para realizar la función para la cual fue diseñada, pero que es capaz de inducir cambios en la estructura terciaria de sus vecinas, transformándolas a su vez en priones. Uno de los ejemplos más notables y recientes de patologías inducidas por priones, es el caso de la encefalopatía espongiforme bovina (enfermedad de las vacas locas) [10].
Uno de los primeros pasos en el problema del plegamiento, se debe a C. Levinthal [11], quien en 1968 demostró que el problema de hallar la configuración terciaria de una proteína necesariamente implica un proceso evolutivo. Para llegar a esta conclusión, observó que si una proteína estuviese integrada por N aminoácidos, y si cada uno de ellos pudiese adquirir, en promedio, un número ν de configuraciones espaciales, entonces el número C de posibles configuraciones tridimensionales está dado por

Si consideramos el caso de una proteína de dimensiones pequeñas, digamos, con N = 100, y suponiendo para el parámetro ν su valor mínimo ν = 2, tendríamos que el número de conformaciones posibles C es,

el cual es número astronómicamente grande. El tiempo necesario para que una proteína con estas características explorase todas las posibles configuraciones tridimensionales hasta encontrar la estructura más estable correspondería a un tiempo mayor que la edad del Universo. Evidentemente esto no ocurre. Las proteínas más pequeñas (N ~ 64 aminoácidos), alcanzan su estructura terciaria en tiempos del orden de milisegundos (t ~ 10-3 seg.), en tanto que proteínas más complejas se pliegan en tiempos no mayores a algunas decenas de segundos (t ~ 101 seg.). Recientemente se ha podido seguir con cierto detalle las etapas del proceso de plegamiento para las unidades básicas que conforman la mayoría de las proteínas, las así llamadas hélices α y hojas β, observándose que estas estructuras se pliegan en tiempos del orden de t ~ 10-6 seg. Este fenómeno, conocido como plegamiento ultrarápido (ultra-fast folding), es una de las líneas más activas en la actualidad en la cinética del proceso [13], [14]. La Paradoja de Levinthal plantea pues la necesidad de que los mecanismos de plegamiento deban poseer un elemento de presión evolutiva que dirija el proceso en una dirección particular y evite una búsqueda aleatoria dentro de todo el espacio de posibles conformaciones.
De acuerdo a lo anterior, el problema del plegamiento de una proteína no sólo consiste en determinar los mecanismos físicos y químicos (si es que existen) que le permitan a la cadena de aminoácidos adquirir una estructura tridimensional específica capaz de llevar a cabo las diversas funciones bioquímicas para las cuales fue diseñada, sino también explicar la estabilidad termodinámica observada para la estructura terciaria y, simultáneamente, sustentar las razones por las cuales este proceso ocurre en tiempos mucho menores que los planteados por la Paradoja de Levinthal. Así dicho, el problema es una tarea inmensa. A lo largo de las siguientes secciones discutiremos someramente los distintos enfoques teóricos que se han propuesto en las dos últimas décadas para tratar de esclarecer este problema.
Para hacer esta revisión autocontenida, la Sección 2 resume brevemente los elementos básicos en la estructura de las proteínas. En la Sección 3 realizamos algunas consideraciones sobre la aplicación del formalismo de la termodinámica de equilibrio al plegamiento de proteínas de dos estados: estructura primaria-estructura terciaria, es decir, sin la existencia de estados intermediarios estables. Finalmente, la Sección 4 repasa las similitudes halladas entre las cadenas heteropoliméricas, los vidrios de espín y las proteínas a fin de generar una teoría microscópica consistente con las observaciones experimentales. También presentamos una enumeración de los resultados más importantes en esta dirección y las dificultades asociadas.
Estructura de las proteínas
Tal como hemos mencionado anteriormente, la estructura primaria de una proteína consiste en una hebra unidimensional conformada por N aminoácidos. La secuencia específica en que estos N aminoácidos están presentes en la cadena lineal está determinada exclusivamente por el ADN y el ARN, y es única para cada proteína. Si tomamos como ejemplo el caso de la molécula del ADN, observamos que existen sólo cuatro tipos distintos de nucleótidos que conforman la doble hélice: guanina (G), citosina (C), adenina (A) y tiamina (T). En contraste, en el caso de las proteínas existen q = 20 tipos distintos de aminoácidos, los cuales, para una cadena de N unidades se pueden combinar de qN maneras distintas, generando un número equivalente de secuencias primarias. Para una proteína integrada por N = 64 aminoácidos, como la quimotripsina inhibidor 2, (CI2), mostrada en la Figura 1, existen en principio 2064 distintas posibles secuencias de aminoácidos. Este número es aún mayor que C, el número de configuraciones posibles para la estructura terciaria, haciendo que el problema de establecer cuál es la secuencia correcta de aminoácidos que permitirá a la macromolécula evolucionar hacia el estado terciario sea complementario al problema del plegamiento y similar a él en cuanto a complejidad. En la sección 4, discutiremos un poco más acerca del papel de las secuencias de aminoácidos en las propiedades de la estructura terciaria.
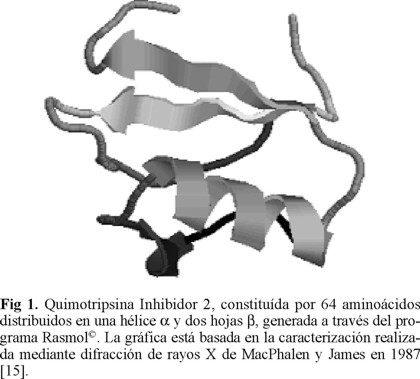
Debido a que las estructuras terciarias no pueden ser predichas hasta el momento bajo formalismo teórico alguno, su determinación se basa en técnicas experimentales, particularmente en dos: la difracción de rayos X y la espectroscopía por resonancia magnética nuclear (RMN). Como dato interesante, mencionaremos que hasta junio del 2003, se habían determinado a través de alguna de esta dos técnicas las secuencias de alrededor de 250,000 proteínas. De estas 250,000 proteinas, se conocen aproximadamente 20,000 estructuras terciarias, entre proteínas, virus y ácidos nucleicos [16]. La Tabla 1 resume estos resultados.

Pese a que existen 20 clases distintas de aminoácidos, todos comparten una estructura química construída con elementos comunes. Cada uno de ellos está integrado por un átomo central de carbono, denominado Cα, cuyas cuatro valencias están ligadas a un grupo amino (NH2), un grupo ácido o grupo carboxilo (COOH), un átomo de hidrógeno (H) y un grupo R llamado residuo, el cual es distinto para cada aminoácido. Es justamente la estructura y composición química del grupo residuo R (en inglés conocido como side chain), lo que establece la diferencia entre cada aminoácido. Para todos los aminoácidos, el grupo residuo R se encuentra ligado al átomo central de carbono Cα, con excepción del aminoácido (Prolina), cuyo grupo residual R está ligado covalentemente tanto al átomo central Cα como al átomo de nitrógeno N del grupo amino [17]. Para los 19 aminoácidos restantes, la estructura general es la siguiente,

Con base en las propiedades físicas de los grupos residuo respecto del solvente en el cual se encuentran inmersos más comúnmente (H2O), es posible definir una clasificación estándar de los 20 aminoácidos conocidos. Esta clasificación los agrupa en tres categorías principales. La primera categoría consiste en aquellos aminoácidos con propiedades hidrofóbicas, es decir, aquellos que presentan interacciones electrostáticas repulsivas hacia las moléculas de H2O. En contraposición, la segunda categoría agrupa a los aminoácidos hidrofílicos o polares, que son aquellos cuya interacción electrostática con las moléculas del solvente es atractiva; y en una tercera categoría se encuentran los aminoácidos cuyo grupo residual tiene carga eléctrica distinta de cero. El aminoácido Gly se considera dentro de una categoría distinta a las ya mencionadas, dado que su grupo residuo consiste solamente en un átomo de hidrógeno y por tanto, exhibe propiedades distintas a las anteriores [18]. En la tabla (2) se enlistan los 20 aminoácidos conocidos, exceptuando el Gly, en relación a las categorías mencionadas.

Ángulos diédricos
Para describir la estructura terciaria de una proteína es posible elegir distintos conjuntos de variables, por ejemplo el conjunto de coordenadas espaciales (x, y, z) para cada átomo. Aunque esta descripción pueda considerarse a primera vista la más natural, es posible definir un conjunto alternativo de coordenadas que nos permita extraer mayor información. En 1963, Ramachandran [19] observó que es posible definir por completo la estructura terciaria mediante el conjunto de ángulos φ y ψ entre los enlaces de cada aminoácido con sus vecinos inmediatos. Un análisis simple muestra que si consideramos una proteína formada por N aminoácidos y definimos como el ángulo φ al ángulo que forma el enlace peptídico entre el aminoácido n - 1 y el aminoácido n y como el ángulo ψ al ángulo que forma el enlace entre el aminoácido n y el aminoácido n + 1, el conjunto de N - 1 ángulos diédricos constituye un conjunto completo de coordenadas generalizadas para la descripción de la estructura terciaria. Sumado a ello, el conjunto de ángulos diédricos demostró jugar un papel relevante en la caracterización de los distintos tipos de estructuras observadas y su funcionalidad.
Estudios experimentales realizados por Morris [20] en 1992 para un conjunto de 300 estructuras terciarias, demostraron que los valores de los ángulos diédricos están restringidos a ciertos subconjuntos. Usando la distribución experimental de los ángulos diédricos observada, es posible definir tres regiones principales en donde se concentran los valores de los ángulos φ y ψ. Estas regiones se denominan region α, región β y región αL. Los nombres asignados para las regiones corresponden a tres clases generales de estructuras, la hélice α, la hoja β y la hélice α izquierda.
Hélices α y Hojas β
El análisis de la estructura de la molécula de mioglobina realizado por Kendrew en 1958 permitió observar una propiedad general de muchas estructuras terciarias que había pasado inadvertida hasta entonces. Kendrew hizo notar que el núcleo de la mioglobina estaba constituído en su enorme mayoría por aminoácidos hidrofóbicos, en tanto que la superficie expuesta al solvente estaba integrada por aminoácidos polares. Esta característica, observada posteriormente para un número considerable de estructuras terciarias, indujo a extraer una idea sobre los mecanismos que utiliza la proteína para generar estructuras termodinámicamente estables.
Partiendo de esta observación, se planteó que el mecanismo principal en el plegamiento de las proteínas es de carácter físico y se debe a la interacción entre los aminoácidos hidrofóbicos e hidrofílicos con las moléculas de H2O, de forma tal que aquellos aminoácidos hidrofóbicos tienden a aglutinarse en el interior de la macromolécula y aquellos hidrofílicos tienden a ocupar la superficie exterior, quedando expuestos a la interacción atractiva con las moléculas del solvente. Pese a que esta idea resulta plausible desde el punto de vista físico y de hecho ocurre para mezclas de ciertos líquidos, por ejemplo agua y aceite, el mecanismo de interacción hidrofóbico-hidrofílico presenta un problema para el caso específico de las proteínas. Para aglutinar a los aminoácidos hidrofóbicos en el núcleo, los átomos Cα correspondientes deben también agruparse en el núcleo. Sin embargo, se sabe que la cadena principal de átomos de carbono Cα es básicamente de carácter polar. Para anular la interacción polar de la cadena de átomos Cα con las moléculas de H2O, se requiere la presencia de puentes de hidrógeno que balanceen esta interacción. La formación de los puentes de hidrógeno conduce a la formación de dos tipos de estructuras en el interior de la macromolécula: las hélices α y las hojas β. Ambas estructuras, conocidas como estructuras secundarias, se caracterizan justamente por la formación de puentes de hidrógeno entre los grupos amino NH2 y los grupos ácido COOH de la cadena principal.
La figura (2) muestra una simulación por computadora de la estructura 3D de la mioglobina, la cual es un ejemplo de una estructura terciaria formada exclusivamente por hélices α. En términos generales, una hélice α contiene 3.6 aminoácidos en promedio por vuelta con un puente de hidrógeno entre el grupo COOH del aminoácido n y el grupo NH2 del aminoácido n + 4. Se han observado variaciones a este modelo de hélice α, en donde los aminoácidos están agrupados con puentes de hidrógeno entre los aminoácidos con periodicidad n + 5 o n + 3 en lugar de n + 4. Estas estructuras se han denominado hélices π y hélice 310, respectivamente. Estas configuraciones se observan en pocas proteínas, debido a que no son energéticamente favorables. Sólo en la hélice α es donde los átomos están empaquetados en forma adecuada para generar una configuración robusta.

La segunda estructura estable formada por puentes de hidrógeno se conoce como hojas β. A diferencia de las hélices α, en donde las interacciones entre aminoácidos es de corto alcance, la formación de una hoja β requiere de interacciones de largo alcance entre aminoácidos. Esta configuración agrupa entre 5 y 10 aminoácidos extendidos aproximadamente en un plano [18]. La mayoría de las proteínas está constituída por combinaciones de estas dos estructuras secundarias, conectadas entre sí mediante cadenas de aminoácidos con forma irregular. Éstas se conocen como hebras de enlace (loop regions) y están constituídas, en promedio por no más de 8 aminoácidos. Debido a que la formación de las hélices α y las hojas β constituyen la respuesta física al problema de la interacción hidrofóbica-polar con el solvente, se encuentran fundamentalmente formando parte del núcleo de la proteína. Las hebras enlace se observan primordialmente en la superifice exterior y su composición está formada básicamente de aminoácidos polares.
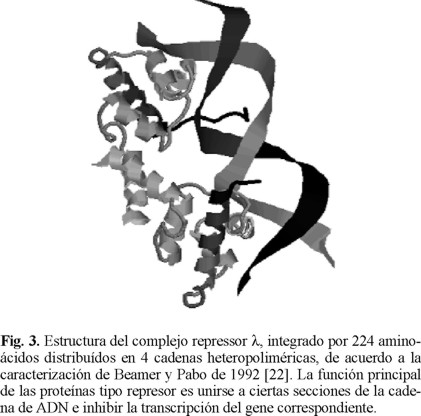
En el siguiente nivel de organización de los aminoácidos, distintas estructuras secundarias se agrupan entre sí para formar lo que se conoce como un dominio. Por convención, se llama dominio a un conjunto de hélices α, hojas β y hebras de enlace que pueden plegarse en forma simultánea e independiente de las partes restantes de la proteína y que además realiza una función específica. Dependiendo de la complejidad de la función realizada por una proteína, ésta puede estar conformada por uno o varios dominios. Por ejemplo, la proteína conocida como Factor de Crecimiento Epitelial (EGF), está integrada por 53 aminoácidos agrupados en un solo dominio. En contraposición, el complejo λ repressor (λ-represor complex), que participa en procesos de enlace con el ADN y en la síntesis de otras cadenas polipeptídicas, se observan dos dominios. El primero formado por dos hojas β y el segundo por dos hélices α (Figura 3).
Una vez establecidos los principales rasgos que caracterizan a las estructuras proteicas, iniciaremos una breve revisión de algunas de las propiedades de la transición estado plegado-estado no plegado para proteínas de dos estados desde el punto de vista de la termodinámica. En particular, haremos énfasis en el papel que juega el calor específico Cp dentro de este esquema y mostraremos que, bajo ciertas condiciones, es posible hallar una expresión analítica para el cambio de la energía libre ΔG del sistema en función de la temperatura T y del calor específico Cp.
Aspectos Termodinámicos
Para considerar al problema del plegamiento de las proteínas dentro de un marco fisicoquímico, es importante tener en cuenta las siguientes hechos,
• Una proteína es un cadena heteropolimérica, integrada por aproximadamente 102 - 104 átomos.
• En el estructura natural o terciaria, los átomos se encuentran ubicados en posiciones espaciales específicas y unívocas, como sucede en un cristal. La diferencia principal con éste último, es que en las proteínas el arreglo de átomos no corresponde a un orden periódico, que sin embargo podemos clasificar como una estructura ordenada.
• Para proteínas pequeñas, el proceso de plegamiento de la estructura primaria es un proceso reversible. Este proceso está gobernado en esencia por las condiciones fisicoquímicas del solvente con el cual interactúa (pH, agentes químicos desnaturalizantes, temperatura, etc).
El proceso de plegamiento de la cadena unidimensional en la estructura terciaria es un mecanismo que implica un incremento en el orden del sistema. En términos de la entropía S, el proceso de plegamiento tiene asociado un cambio de entropía ΔS negativo, lo cual es termodinámicamente poco favorable. Para que la estructura terciaria corresponda al estado termodinámicamente más favorable es necesario que este efecto entrópico sea compensado por una contribución negativa en el cambio de la energía interna ΔE del sistema, de forma que el cambio de la energía libre ΔF del sistema sea negativo [23], dado que,

La contribución energética ΔE debe provenir del reacomodo de los átomos dentro de la macromolécula y consecuentemente, de la redistribución de las interacciones presentes, a fin de alcanzar un estado extremal. Las interacciones principales de carácter no covalente (es decir, aquellas que no implican la ruptura de los enlaces covalentes entre los átomos Cα de la estructura primaria), son las interacciones de Van Der Waals, las interacciones entre grupos con carga eléctrica, los puentes de hidrógeno entre grupos polares y la llamada interacción hidrofóbica entre grupos no polares. Estudios experimentales [24], [25] apuntan a que la contribución más significativa en la estabilización de la estructura terciaria proviene de la interacción hidrofóbica entre los aminoácidos no polares y las moléculas de agua. Esta idea se ve reforzada por la observación experimental de que el núcleo de la estructura terciaria está formado por un cúmulo de aminoácidos no polares, en forma similar a lo que ocurre cuando se trata de disolver una sustancia no polar en agua. La poca solubilidad en agua observada para las sustancias polares indica que la transferencia de moléculas del soluto al solvente requiere un gasto en energía para llevarse a cabo, esto es, el cambio de la energía ΔG del proceso de mezclado debe ser positivo. Asimismo, se ha hallado que el cambio de la entalpía ΔH asociado a la mezcla de estas sustancias en agua es prácticamente cero a temperatura ambiente [26], [27]. Por tanto, dado que el cambio en la energía libre está asociado con un cambio en la entropía a través de la relación,

se puede inferir que el ΔG > 0 observado, está asociado a un ΔS < 0 significativo para el proceso de mezclado. Los valores experimentales para ΔG, ΔH, ΔS y ΔCp del mezclado de diversas sustancias no polares en agua se consignan en la tabla (3) [28].

Dado que un proceso con un cambio ΔS < 0 es termodinámicamente poco favorable, la observación experimental anterior implica que el proceso de mezclado de grupos no polares en agua es un proceso que debe ocurrir paralelamente a un cambio en la entropía positivo en el solvente. En otros términos, que la presencia del soluto induce un mayor orden en las moléculas del solvente [29]. Esta afirmación está en acuerdo con la observación experimental de un incremento en el calor específico Cp del agua en presencia de solutos no polares [30]. Inpendientemente de que ésta sea la explicación correcta al decremento observado de la entropía en el proceso de mezclado, el punto relevante aquí es mostrar que el mecanismo de mezclado de sustancias no polares en agua es un proceso termodinámicamente no favorable. Para evitar esta situación, el solvente expulsa a los grupos no polares, ocasionado la formación de agregados. Este mecanismo fue propuesto por primera vez por Kauzmann en 1959 [31], y constituye la explicación termodinámica de la interacción hidrofóbica entre los aminoácidos que constituyen a las proteínas y las moléculas de H2O. En la actualidad, la propuesta de Kauzmann es considerada como uno de los elementos más importantes en el plegamiento de los complejos proteicos.
Una vez establecido uno de los principios básicos que rigen la dinámica del plegamiento, procederemos a analizar la aplicación del formalismo termodinámico a este tipo de sistemas y mostraremos que conclusiones pueden obtenerse de él. El sistema más simple que puede considerarse es una proteína que sólo puede adquirir dos estados posibles: el estado desnaturalizado (cadena lineal de aminoácidos sin estructura tridimensional) y el estado terciario, en donde la hebra se ha plegado para adquirir una estructura tridimensional específica. Aunque podría pensarse que dada la complejidad de las macromoléculas biológicas esta situación es sólo de interés académico, en realidad no es el caso. En 1990, Ferhst [32] observó que para la proteína quimiotripsina inhibidor 2, (CI2), la cual es una proteína pequeña constituída por 64 aminoácidos, esto es justamente lo que ocurre. En este caso, la proteína sólo puede adquirir dos estados estables: el estado desnaturalizado D, y el estado nativo N. Además de lo anterior, el plegamiento de CI2 es un proceso reversible.
Para este tipo de sistema, el mecanismo de plegamiento puede plantearse como una cinética de primer orden de acuerdo a la reacción siguiente

Utilizando esta analogía con las reacciones químicas, en la cual una especie A se transforma en una especie B, es posible aplicar el formalismo termodinámico correspondiente y por tanto, plantear el conjunto de relaciones termodinámicas para la transición D ⇄ N. Estas relaciones son,

en donde k representa la tasa de conversión del estado D al estado N. Es importante mencionar que la justificación del planteamiento anterior yace en el hecho de que la proteína sea, efectivamente, un sistema de dos estados, es decir, que no existan estados intermediarios estables en el proceso de plegamiento2. La verificación de este hecho ocurre cuando las propiedades termodinámicas que se derivan de las relaciones (6) corresponden a las observadas experimentalmente. En la práctica esto es posible sólo en experimentos de desnaturalización inducida por temperatura, en los cuales la variable extensiva asociada a la temperatura T es la entalpía H del proceso, la cual puede medirse con suficiente precisión a través de las técnicas de microcalorimetría de barrido [33].
Estudios experimentales de desnaturalización de proteínas inducidos por temperatura muestran que, en el caso de proteínas globulares cuya masa molecular M es menor que 20 kDa, la desnaturalización va acompañada de un incremento significativo en la curva de absorción de calor. En términos termodinámicos, el calor absorbido del sistema está asociado al calor específico Cp de la macromolécula, por tanto en este caso, se observa un pico en la curva Cp a medida que la temperatura T se incrementa. La técnica de microcalorimetría de barrido desarrollada por Privalov [34] permite medir con suficiente precisión el calor específico Cp en función de la temperatura. Una vez conocido el Cp en un cierto intervalo de temperaturas T1 - T2, es posible utilizar la relación

para calcular la forma funcional de la entalpía H en función de la temperatura T mediante una integración directa de (7), esto es,

Asimismo, es posible determinar no sólo la entropía S del sistema a través de la relación,
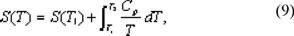
sino también la energía libre de Gibbs G del sistema, a partir de la entalpía H y la entropía S.

El planteamiento anterior muestra que es posible determinar la forma funcional de la energía libre de Gibbs G en función de la temperatura si conocemos con suficiente precisión el comportamiento del calor específico Cp en el intervalo de temperaturas de interés. La relevancia de este hecho ha sido reconocida desde el trabajo pionero de Hutchens [35] y continuado en la actualidad por diversos grupos de investigación que han dedicado esfuerzos considerables al diseño y perfeccionamiento de técnicas experimentales para medir el Cp o, más apropiadamente el ΔCp de la transición D ⇄ N.
Es importante resaltar que el esquema anterior no resuelve el problema de la predicción de la estructura terciaria a partir de la secuencia inicial de aminoácidos ni el problema de la paradoja de Levinthal, aspectos centrales en un análisis completo del problema del plegamiento. Sin embargo, su relevancia yace en la posibilidad de calcular teóricamente el valor experimental observado para ΔG en la transición D ⇄ N en diversas proteínas, como veremos a continuación. Experimentalmente se observa que la estabilización de la estructura terciaria ocurre dentro del intervalo 40 kJmol-1 < ΔG < 50 kJmol-1 [36] en proteínas de un solo dominio. Dado que es posible inducir la desnaturalización de una proteína mediante la adición de agentes químicos como la urea o bien mediante efectos de presión, entre otros, inicialmente se consideró que el incremento de la capacidad calorífica podría, en principio, depender del proceso de desnaturalización utilizado, y por tanto, restringir la validez del esquema anterior. Sin embargo este no es el caso. Diversos experimentos realizados con distintos agentes desnaturalizantes para detectar diferencias en el ΔCp han arrojado resultados negativos sobre esta cuestión [37]. Si existe una diferencia entre los estados desnaturalizados que dependa del proceso utilizado, esta diferencia no es relevante desde el punto de vista termodinámico.
Una vez establecida la relevancia del calor específico dentro de una descripción termodinámica de la transición D ⇄ N, mostraremos cómo dependen los potenciales termodinámicos H y S en función de Cp y T y calcular explícitamente la forma funcional del ΔG. Para ello, supondremos que conocemos con suficiente precisión el valor del calor específico Cp en el intervalo de temperaturas TG - T, en donde TG es la temperatura del punto medio de la transición D ⇄ N. Es decir, TG corresponde a la temperatura en donde la energía libre G del estado D es igual a la energía libre G del estado N. Si la transición ocurriese en forma instantánea, entonces ocurriría exactamente en la temperatura TG. Dado que en los procesos reales la fractura de la estructura terciaria no ocurre instantáneamente, la transición D ⇄ N sucede dentro de un intervalo de temperaturas. La temperatura media en este intervalo es justamente TG. Por construcción, TG satisface la ecuación siguiente,

de donde se deduce que
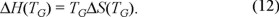
Con esto en mente e integrando la ecuación (7) en el intervalo TG - T, obtenemos

Para calcular el cambio de entropía ΔS en el mismo intervalo de temperaturas, observemos que

por tanto, integrando (14) en el intervalo de interés, obtenemos

Las relaciones (13) y (14) para los potenciales H y S son de carácter general. En el caso de que el ΔCp sea una constante, obtenemos una función lineal para el cambio de la entalpía en términos de T,
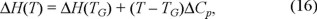
y una dependencia logarítmica para el cambio de la entropía,

en donde hemos utilizado la ecuación (12).
Diversos estudios realizados en 1988 por Privalov y Gill [38] para diversas proteínas globulares como la lisosima, la ribonucleasa A y la mioglobina, entre otras, han mostrado que el ΔH de la transición D ⇄ N es una función lineal de la temperatura, en congruencia con las mediciones realizadas por calorimetría de barrido en donde se muestra que ΔCp es constante.
Para concluir esta sección, calcularemos la forma funcional de ΔG en función de la temperatura T y la forma de ΔCp(T), partiendo de los potenciales H y S calculados previamente [25], [28]. Con base en la ecuación (10), es posible escribir los cambios de los potenciales termodinámicos en el intervalo TG - T de acuerdo con,

Sustituyendo (13) y (15) en (18), obtenemos

Agrupando términos y utilizando la ecuación (11), obtenemos para ΔG(T),

Para el caso en que ΔCp es una constante en el intervalo de integración, se obtiene finalmente que,

La característica más importante de este resultado es que, a diferencia de la entalpía y la entropía que son funciones crecientes de T, el ΔG es una función que exhibe un valor máximo. Si llamamos TS a la temperatura a la cual el ΔG tiene un valor extremal, vemos que se satisface la siguiente ecuación,

que para el caso de ΔCp constante corresponde a,

El desarrollo anterior muestra que es posible calcular explícitamente los potenciales termodinámicos H y S, y, en consecuencia, la energía libre G para el proceso de desnaturalización de una proteína de dos estados. Asimismo es posible determinar la temperatura en donde el cambio ΔG exhibe un valor extremal, el cual es único y corresponde a la diferencia entre las energías del estado desnaturalizado y de formación de la estructura terciaria. Estos resultados, publicados previamente en las referencias citadas, han sido verificados experimentalmente para diversas proteínas globulares, en las cuales la desnaturalización ocurre en dos etapas. Asimismo constituyen una base de referencia importante en la construcción de modelos microscópicos para la dinámica del plegamiento y sus problemas asociados.
Aspectos microscópicos: vidrios de espín y heteropolímeros
En la sección anterior hemos discutido brevemente algunas de las características termodinámicas que caracterizan la transición D ⇄ N para el caso de proteínas de dos estados. Por construcción, el enfoque termodinámico nos permite dilucidar algunas de las características más importantes de los estados de equilibrio de un sistema, en este caso, del estado nativo de la macromolécula. Sin embargo, este enfoque resulta limitado cuando se desea abordar el problema de la dinámica y cinética del plegamiento. Para estar en posibilidades de abordar este problema, se han propuesto diversos modelos microscópicos que toman en cuenta las interacciones entre los diversos aminoácidos. La complejidad de dichos modelos reduce en gran medida su aplicabilidad a sistemas reales y hacen que la posibilidad de hallar soluciones analíticas sea limitada, salvo en el caso de sistemas simples y aún con el uso extensivo de métodos computacionales. Pese a la dificultad intrínseca del problema, ha sido posible extraer algunas respuestas parciales. En esta sección revisaremos brevemente los avances más importantes en esta dirección.
Uno de los primeros pasos en el desarrollo de una teoría microscópica del plegamiento proviene de la analogía observada entre el plegamiento de un proteína y ciertas transiciones de fase observadas para las cadenas homopoliméricas en solución. En los trabajos clásicos de Flory [5], DeGennes [6] y Volkenstein [39], se establece que una cadena homopolimérica unidimensional puede colapsarse para formar una estructura globular compacta, en donde las fluctuaciones de la densidad se han reducido a su mínimo valor. Esta transición, conocida en la literatura como transición hebra-glóbulo (coil-globule transition) ha sido discutida analíticamente por Lifshitz et al [40] y, más recientemente, por Grosberg y Kuznetsov [41] para cadenas homo y heteropoliméricas, respectivamente. El resultado más importante que se desprende de los estudios realizados por Lifshitz et al, es que la transición hebra-glóbulo constituye una transición de fase en términos estrictos, es decir, en el límite termodinámico, se observa una discontinuidad en las funciones termodinámicas del sistema.
La limitante más importante de los trabajos anteriores radica en que se supone que las cadenas hetero y homopoliméricas analizadas poseen un orden secuencial de los aminoácidos, en tanto que en las proteínas reales se observa que la secuencia de aminoácidos es, en primera aproximación, de carácter aleatorio. Para tomar en cuenta el efecto del aparente desorden en las secuencias de aminoácidos, Bryngelson y Wolynes [42] han propuesto que una proteína, al estar constituída por aminoácidos de distintas clases y por tanto con interacciones energéticas de distintos tipos, debe poseer propiedades similares a las observadas en los vidrios de espín. Este enfoque, conocido como teoría del paisaje energético (energy landscape theory) supone que, en forma análoga en que una macromolécula puede adquirir un número enorme de configuraciones tridimensionales, también es posible definir un número astronómico de posibles secuencias de los aminoácidos. Dado que muchas de estas secuencias son similares a la secuencia específica que producirá el plegamiento, la energía de esta secuencias es similar a la energía de la estructura terciaria, generando que el paisaje energético de la macromolécula, es decir, el gráfico de la energía del sistema en función de la configuración, presente un aspecto rugoso, con múltiples mínimos locales separados por barreras energéticas y un mínimo global correspondiente a la configuración nativa (Fig. 4).

En la teoría de los vidrios de espín, los espines de los distintos átomos pueden apuntar en direcciones arbitrarias. A medida que el sistema se enfría, los espines se reacomodan tratando de encontrar su configuración de mínima energía. Dado que existen múltiples interacciones, no sólo con los vecinos inmediatos sino también de largo alcance, se produce una situación de conflicto para el sistema al tratar de encontrar la configuración energética más favorable, dado que en principio, dos configuraciones similares tendrán energías similares y es difícil para el sistema decidir cuál configuración adoptar. Este fenómeno es conocido como frustración, y constituye la base de la teoría del paisaje energético. Cuando un sistema físico presenta el fenómeno de la frustración, el estado de mínima energía global no es más estable que los estados con energías similares (Fig. 5).

Considerando la analogía de los vidrios de espín como sistemas altamente frustrados con las secuencias aleatorias de aminoácidos observadas en las proteínas, Bryngelson y Wolynes han propuesto que las proteínas cuyas secuencias sean aleatorias deben ser sistemas que presentan dificultades para hallar su configuración energética más estable. Pese a que las proteínas parecen estar integradas por secuencias aleatorias de aminoácidos, se comportan de forma completamente distinta a los vidrios de espín en términos de alta frustración. Por ejemplo, en el caso de proteínas de dos estados, la transición entre el estado desnaturalizado y el estado terciario ocurre en forma directa, sin la rugosidad prevista por la teoría del paisaje energético.
Esta situación ha sugerido la idea de que las proteínas reales deben ser sistemas con frustración mínima y por tanto, ser el resultado de un proceso evolutivo en donde se han seleccionado las secuencias de aminoácidos que mejor resuelven el problema de la frustración. Este hecho introduciría una componente netamente biológica al proceso, no necesariamente fisicoquímica. La presencia de la frustración en un sistema impone una estructura particular al paisaje energético. Dado que en un sistema de este tipo existen numerosos estados cuya energía es similar al estado base, el paisaje energético debe poseer la forma de un embudo con múltiples mínimos locales separados por igual número de barreras energéticas, en donde el extremo del embudo corresponde al estado energéticamente más favorable. Dentro de este esquema, sin embargo, existen diversas posibilidades dependiendo del grado de frustración. Para un sistema altamente frustrado, no existe la posibilidad de un mínimo global, dado que todas las configuraciones poseen energías similares. Para el caso de sistemas con frustración mínima, o dicho de otra manera, con frustración optimizada, el paisaje energético deberá poseer una forma de embudo, similar a la descrita anteriormente.
Uno de los resultados más notables de las teoría del paisaje energético de Bryngelson y Wolynes es el concepto de transición por congelamiento de estados de las cadenas heteropoliméricas [45], es decir, la transición de una cadena heteropolimérica entre dos estados termodinámicamente distintos. En esta transición, el número de estados O accesibles al sistema cambia de orden de magnitud, de O ~ N, a O ~ exp N. Este fenómeno se observa en los vidrios de espín y es causado por el fenómeno de la frustración. Cuando esta transición fue predicha, primero fenomenológicamente por Bryngelson y Wolynes [42] y después dentro de un esquema microscópico por Shakhnovich y Gutin [45], [46], parecía ser una respuesta satisfactoria para el plegamiento de las proteínas, dado que en este modelo, existe la posibilidad de un estado termodinámico congelado, el cual podría identificarse, en principio, con el estado tercario de las proteínas.
Tanto el enfoque fenomenológico de Wolynes, como el enfoque microscópico de Shakhnovich utilizan explícita o implíctamente la idea de que las secuencias de aminoácidos tienen un carácter esencialmente aleatorio. Sin embargo, de acuerdo a las consideraciones anteriores, éste no necesarimente es el caso. Los principales obstáculos que se observan dentro de este esquema son los siguientes [47],
• Sólo existe una secuencia aleatoria que se congela. Sin embargo, la configuración tridimensional correspondiente a este estado también es aleatoria. Este resultado es inaceptable desde el punto de vista biológico, ya que una proteína tiene una estado nativo único, incluso cuando la secuencia de aminoácidos ha sido modificada genéticamente.
• El estado base de la cadena heteropolimérica aleatoria es estable, sin embargo, los estados próximos a él sólo difieren energéticamente por un factor
pese a tener configuraciones terciarias completamente distintas. Dada esta similitud, estos estados funcionan como trampas energéticas, haciendo que el tiempo de plegamiento se incremente en varios órdenes de magnitud.
Para resolver el problema presentado por las secuencias aleatorias, Pande, Grosberg y Tanaka [47] han propuesto extender el formalismo anterior utilizando secuencias de aminoácidos mínimamente frustradas, las cuales, como se mencionó anteriormente, están asociadas con paisajes energéticos de tipo embudo y distan de ser aleatorias. En términos técnicos, el enfoque utilizado por Pande et al, consiste en esencia, en calcular la energía libre de F del sistema mediante un promedio estadístico sobre un espacio de secuencias ES, el cual corresponde a todas las posibles cadenas lineales que pueden formarse con N aminoácidos y q distintos tipos de ellos, y sobre un espacio de configuraciones EC, el cual corresponde a las posibles estructuras terciarias que pueden formarse con las secuencias mencionadas, el cual como vimos en la Introducción, está conformado por aproximadamente eN elementos. La energía libre F del sistema se puede representar entonces por,

en donde la notación 〈x〉y denota un promedio estadístico de la variable x sobre un ensamble en el espacio y. La ecuación (24) indica que el promedio se realiza en primer lugar sobre las posibles secuencias a fin de encontrar las más óptima desde el punto de vista de la frustración y, en segundo lugar, se realiza el promedio sobre las posibles configuraciones tridimensionales accesibles para dicha secuencia. Para realizar tales promedios, se utiliza la función de partición Z definida sobre ambos espacios y su relación con la energía libre F, de acuerdo a la relación,
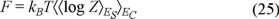
en donde kB denota la constante de Boltzmann y T es la temperatura del sistema.
La aparente simplicidad de la ecuación (25) es engañosa. Para estar en posibilidades de calcular la energía libre F del sistema bajo este esquema, se requiere conocer la función de partición Z del sistema, la cual depende a su vez de la función de Hamilton H. Para poder determinar entonces Z se requiere de un modelo de las interacciones, tanto de corto como de largo alcance entre los diversos aminoácidos. Debido a la complejidad de las distintas interacciones presentes, esta posibilidad se encuentra lejos de concretarse, por lo menos hoy en día.
Así planteado, la magnitud del problema es formidable. Basta decir que en una proteína típica existen alrededor de N ~ 102 aminoácidos y que existen q = 20 clases distintas de ellos. La función de Hamilton más simple debería contener del orden de N términos de interacción a primeros vecinos, sin tomar en cuenta que gran parte de los mecanismos estabilizadores de las estructuras secundarias y terciarias proviene de interacciones de largo alcance y que la dimensión de los espacios S y C crece exponencialmente con la dimensión N del sistema. Aún estando en posibilidades de establecer una función de Hamilton apropiada, el cálculo de la función de partición Z presenta sus propias dificultades. Como ejemplo de ello recordemos que en el caso de un modelo simplificado para un vidrio de espín mediante el modelo de Ising, resulta imposible calcular la función de partición del sistema en tres dimensiones en forma exacta. Pese a todos estos obstáculos, Pandé et al han podido obtener algunos resultados parciales, utilizando una combinación de simulaciones numéricas y aproximaciones para modelar la función de Hamilton. Entre los más destacados se encuentra la predicción teórica de la temperatura Tf a la cual el sistema exhibe una transición de fase, la cual depende, entre otros parámetros de la secuencia de aminoácidos elegida. El identificar a esta temperatura con la temperatura real del plegamiento en las proteínas es, como mencionan los mismos autores, algo prematuro, y que deberá ser evaluado minuciosamente a la luz de las aproximaciones realizadas y confrontada con experimentos venideros.
Consideraciones Finales
El problema de la predicción del estado nativo de las proteínas a partir de la secuencia inicial de aminoácidos es un problema que ha permanecido abierto en las dos últimas décadas. En este trabajo hemos pretendido realizar una revisión breve sobre los aspectos físicoquímicos más relevantes de las teorías que se han propuesto en las últimas dos décadas para abordar el problema. Nos hemos centrado fundamentalmente a aquellos aspectos de mayor interés para los físicos y por razones de espacio hemos dejado de lado dos enfoques paralelos a los aquí presentados: las simulaciones numéricas utilizando el Método de Monte Carlo y la Dinámica Molecular y, en segundo lugar, pero no por ello menos importante, el tratamiento de los aspectos cinéticos mediante el uso de la Ecuación Maestra y los procesos estocásticos. Debido a la dificultad práctica de calcular o medir las probabilidades de transición entre estados, consideramos que este último enfoque sólo podría proveer de respuestas parciales, en el mejor de los casos. En nuestra opinión, el plegamiento de las proteínas constituye uno de los retos más importantes y de mayor envergadura que afronta en la actualidad la Física, la Química y la Biología, no sólo desde el punto de vista de la generación de ciencia básica, importante per se, sino también por la enorme diversidad de las aplicaciones biomédicas y tecnológicas que apenas alcanzamos a vislumbrar. La colaboración verdaderamente interdisciplinaria sea quizás el vehículo que proporcione la respuesta a tan interesante cuestión en los años venideros.
Uno de los autores (L. Olivares-Quiroz) expresa su agradecimiento al CONACyT-SNI por el financiamiento recibido. Se agradece a E. Vázquez Contreras del Instituto de Química de la UNAM y a M. Costas Basin de la Facultad de Química de la UNAM por sus comentarios y sugerencias.
Referencias
1. Pauling, L.; Corey, R.B. Branson, H.R. PNAS USA. 1951, 37, 205-211. [ Links ]
2. Watson, D.J.; Crick, F.C.H. Nature 1953, 171, 740. [ Links ]
3. Kendrew, J.C. Nature 1958, 181, 662-666. [ Links ]
4. Flory, P.J. Principles of Polymer Chemistry. Cornell University, Ithaca, 1953. [ Links ]
5. Flory, P.J. Statistical mechanics of chain molecules. Interscience, New York, 1969. [ Links ]
6. de Gennes, P.G. J. Phys. (Paris) 1976, 37, 1445. [ Links ]
7. Anfinsen, C.B.; Haber, E.; Sela, M.; White F.W. PNAS USA. 1961, 47, 1309-1314. [ Links ]
8. Dobson, C.M. Trends Biochem. Sci. 1999, 24, 329-332. [ Links ]
9. Dobson, C.M. Philos. Trans. R. Soc. B. 2001, 356, 133-145. [ Links ]
10. Chanez-Cárdenas, M.E.; Cisneros-Armas, D.; Del Pozo-Yauner, L.; Becerril, B.; Fernandez-Velasco. Mensaje Bioquímico 2002, 26. [ Links ]
11. Levinthal, C. J. Chim. Phys. 1968, 65, 44-45. [ Links ]
12. Lifshitz, I.M.; Grosberg, A.Y.; Khokhlov, A.R. Rev. Mod. Phys. 1978, 50, 683. [ Links ]
13. Hochtrasser, R.M. en Protein Folding and Misfolding. European Molecular Biology Organization Symposium, Italy, 2003. [ Links ]
14. Ferguson, N.; Johnson, C.M.; Macias, M.; Oshenkinal, H.; Fersht, A. PNAS USA. 2001, 98, 13002-13007. [ Links ]
15. MacPhalen, C.A.; James, M.N. Biochemistry 1987, 26, 261. [ Links ]
16. Weissing, H., Shindyalov, I.N.; Bourne, P.E. Nucleic Acids Research 2000, 28, 235-242. [ Links ]
17. Creighton, T.E. Proteins: Structures and Molecular Properties. Second Edition. W.H. Freeman Co. 1993. [ Links ]
18. Branden, C.; Tooze, J. Introduction to Protein Structure. Second Edition Garland Publishing Co. 1999. [ Links ]
19. Ramachandran, G.N.; Ramakrisnan, C.; Sasisekharan, V. J. Mol. Biol. 1963, 7, 95-99. [ Links ]
20. Morris, A.L.; MacArthur, M.W.; Hutchinson,E.G.; Thorton, J.M. Proteins: Struc. Funct. Genet. 1992, 12, 345-364. [ Links ]
21. Watson, H.C.; Kendrew, J.C. Prog. Stereo Chem. 1969, 4, 229. [ Links ]
22. Beamer, L.J.; Pabo, C.O. J. Mol. Biol. 1992, 227, 177. [ Links ]
23. García-Colín, S.L. Introducción a la Termodinámica Clásica. Edit. Trillas. 4a. Edición. México D.F. 1989. [ Links ]
24. Hollecker, M.; Creighton, T.E. Biochim.Biophys. Acta. 1982, 701, 395. [ Links ]
25. Creighton, T.E. J. Phys. Chem. 1985, 89, 2452. [ Links ]
26. Baldwin, R.L. PNAS USA. 1986, 83, 8060. [ Links ]
27. Gill, S.J.; Wadso, I. PNAS USA. 1976, 73, 2955. [ Links ]
28. Creighton, T.E. Protein Folding. W.H. Freeman and Company, 1992. [ Links ]
29. Frank, H.S.; Evans, M.V. J. Chem. Phys. 1945, 13, 507. [ Links ]
30. Edsall, J.T. J. Am. Soc. 1935, 57, 1506. [ Links ]
31. Kauzmann, W. Adv. Protein Chem. 1959, 14, 1. [ Links ]
32. Fersht, A.R. Structure and Mechanism in Protein Science. First Edition. New York: W.H. Freeman & Co. 1999. [ Links ]
33. Privalov, P.L.; Potekhin, S.A. Methods Enzymol. 1986, 134, 4. [ Links ]
34. Privalov, P.L. Adv. Protein. Chem. 1979, 33, 167. [ Links ]
35. Hutchens, J.O.; Cole, A.G.; Stout, J.W. J. Biol.Chem. 1969, 244, 25-32. [ Links ]
36. Pfeil, W.; Privalov, P.L. Biophys.Chem. 1976, 4, 41. [ Links ]
37. Pfeil, W.; Bychkova, V.E.; Pititsyn, O.B. FEBS Letters 1986, 189, 287. [ Links ]
38. Privalov, P.L.; Gill, S.J. Adv. Protein Chem. 1988, 39, 193. [ Links ]
39. Volkenstein, M. V. Configurational Statistics of Polymer Chains. Moscow. InterScience, New York, 1963. [ Links ]
40. Lifshitz, I.M.; Grosberg, A.Y.; Khokhlov, A.R. Rev. Mod. Phys. 1978, 50, 683. [ Links ]
41. Grosberg, A.; Kuznetsov, D.V. Macromolecules 1992, 25, 1970. [ Links ]
42. Bryngelson, J.D.; Wolynes, P. G. J. Phys.Chem. 1989, 93, 6902. [ Links ]
43. Dill, K.A.; Chan, H.S. Nat. Struct. Biol. 1997, 4, 1. [ Links ]
44. Chan, H.S.; Dill, K.A. Proteins: Struc. Funct. Gen. 1988, 30, 2-33. [ Links ]
45. Shakhnovich, E.I.; Finkelstein, A.V. Biopolymers 1989, 28, 1967. [ Links ]
46. Shakhnovich, E.I.; Gutin, A. Biophys.Chem. 1989, 34, 187 [ Links ]
47. Pande, V.S.; Grosberg, A.Yu.; Tanaka, T. Rev. Mod. Phys. 2000, 72, 259. [ Links ]
Notas
1 Los sitios activos de una proteína son regiones espaciales en donde se ensamblan otras proteínas o ácidos nucleicos a fin de realizar una función bioquímica particular.
2 Se ha observado que las proteínas de masa molecular pequeña (< 20 kDa) son sistemas de dos estados. 1 Da equivale a 1.65 × 10-24 g.

