




![Photochemistry of 7-Alcoxy and Thioalcoxy-3,3-dimethoxybicyclo[2.2.2] oct-5-en-2-one. Sequence 1,3-acyl shift-decarbonylation reaction](/img/en/next.gif)





Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de la Sociedad Química de México
Print version ISSN 0583-7693
Rev. Soc. Quím. Méx vol.47 n.3 Ciudad de México Jul./Sep. 2003
Investigación
Correlación electrónica fuerte: sistemas con electrones 4f y 5f
Ernesto Chigo-Anota1 y Juan Francisco Rivas Silva*,2,3
1 Posgrado en Ciencias Químicas-Facultad de Ciencias Químicas, Blvd. 14 Sur 6301, 72570, Puebla 72570, Pue., México. Benemérita Universidad Autónoma de Puebla. E-mail: echigoa@sirio.ifuap.buap.mx
2 Instituto de Física "Luis Rivera Terrazas", Apdo. Postal J-48, 72570, Puebla, México. Benemérita Universidad Autónoma de Puebla. E-mail: rivas@sirio.ifuap.buap.mx
3 Instituto Nacional de Astrofísica, Óptica y Electrónica, Coordinación de Óptica. Apdo. Postal 51 y 216, Puebla 72000, Pue., México.
Recibido el 24 de enero del 2003
Aceptado el 22 de agosto del 2003
Resumen
Estudiamos el concepto de correlación electrónica fuerte (CEF) presentada en materiales sólidos tales como los superconductores a alta Tc y los sistemas de fermiones pesados (FP) que contienen iones de Cerio con electrones 4f y iones de Uranio o Neptunio con electrones 5f y compuestos de metales de transición. Dicho efecto ha sido objeto de estudio en teorías modernas y complicadas matemáticamente, por ciertos grupos de investigadores a nivel mundial y no fácilmente aplicadas a programas computacionales.
Palabras clave: Correlación electrónica fuerte, fermiones pesados.
Abstract
We try to explain the strong electronic correlation (SEC) concept which is presented in solid materials such as superconductors at high Tc and the heavy fermion (HF) systems that contain Ce ions with 4f electrons and U or Np ions with 5f electrons and transition metals compounds. The effect has been matter of study in modern and complex new theories by certain groups of researchers they are not easily applied to computational program.
Keywords: Strong electronic correlation, heavy fermions.
Introducción
El Hamiltoniano de un sistema de muchos electrones está dado por (en unidades atómicas h = m = e =1):

donde V es un potencial local externo tal como el campo del núcleo. Resolver el anterior Hamiltoniano ha sido un gran problema en Química Cuántica (QC) y en Física del Estado Sólido (FES). La interacción de Coulomb en el último término hace que el Hamiltoniano se dificulte al tratar de resolverlo.
Ahora bien, la correlación electrónica [1] (como consecuencia de esta interacción coulombiana) ha sido objeto de estudio en ambas ramas: la Química Cuántica Computacional (en adelante QCC) y por la Física del Estado Sólido Computacional (en adelante FESC), debido al comportamiento inadecuado que se tiene cuando modelamos sistemas (ya sean a nivel atómico, molecular o sólidos) y estudiamos su estructura electrónica a nivel teórico (cálculos de primeros principios o ab-initio) en lo que conocemos por simulación computacional [2]. Esto como consecuencia de que en la aproximación Hartree-Fock (en adelante HF) [3] no remueve completamente la autointeracción (repulsiones coulombianas entre los mismos electrones), es decir, se elimina en forma promediada la interacción entre los electrones.

En la ec. (2), el primer término representa la interacción interelectrónica dos veces y es la energía de los orbitales. El segundo término nos indica que para tomar la energía total del átomo tenemos que restar las repulsiones electrónicas, la cual son tomadas en una forma promediada. Jij es una integral de Coulomb, dado que es igual a la energía electrostática de repulsión entre un electrón con una función densidad de probabilidad |gi(i)|2 y un electrón con una función densidad de probabilidad |gj(j)|2.
Por el lado de la teoría DFT[4], encontramos la aproximación LDA (Local Density Approximation, donde la densidad en el potencial de intercambio-correlación del gas de electrones es reemplazado por la densidad local del sistema real), construida análogamente a la teoría del campo medio, la cual toma en una manera promedio la autointeracción entre los electrones, como consecuencia de dicha teoría de campo medio.
El LDA es una aproximación de un electrón (o aproximación del electrón independiente, siendo esto un prerrequisito para aquellos sistemas que incluyen efectos de correlación) con un potencial independiente del orbital (o independiente de la ocupación del orbital).
Además, toma en cuenta la interacción de intercambio en una manera parcial, debido a que el número de electrones con diferente espines son iguales (N↑ = N↓). El LSDA es el que toma en cuenta este intercambio adecuadamente, pero aun así falla.
Por otro lado, en la teoría DFT tenemos,

Los dos primeros términos son análogos a la expresión de la energía total del átomo en la teoría HF. Por tal motivo, lo mencionado arriba de remover en forma promediada la interacción electrónica en el LDA. Aquí, se toma en cuenta ya dichos efectos de correlación-intercambio existiendo varias aproximaciones como el antes mencionado LDA, LSDA (Local Spin Density Approximation), GGA (Generalized Gradient Approximation) y GGS (Gradient -corrected LSDA).
Sabemos que, por el lado, de la QC, tenemos a la teoría HF la cual normalmente está contenida en los programas computacionales GAUSSIAN [5] (enfocado a sistemas moleculares y atómicos) y el tratamiento para sistemas sólidos la encontramos en los programas CRYSTAL [6].
En la FES encontramos a la teoría de funcionales de la densidad (DFT) que podremos encontrar en una variedad de programas computacionales, tales como el CASTEP [7], ESOCS [7], LMTO-ASA [8], FP-LAPW [9], etc., y el mismo Gaussian contiene dicha teoría.
Nuestro enfoque está centrado en sistemas compuestos de fermiones pesados (proveniente del hecho de que γ (coeficiente de Sommerfeld del calor específico) sea del orden de Jmol−1K−2 en comparación con metales simples que es del orden de mJmol−1K−2, implicando esto a su vez una gran masa efectiva el cual es varios cientos de veces la masa del electrón libre, en adelante FP)[10] que contienen iones de Cerio (electrones 4f y cuyo material de interés es el ternario CeFeGe3 [11]), Uranio o Neptunio (electrones 5f cuyo sistemas de interés son el UO[12] y USb[13] que se pueden encontrar en las citas mencionadas), además de dar a conocer los niveles de correlación en los cuales normalmente se trabaja cuando tratamos de describir adecuadamente propiedades de materiales sólidos.
Definición y niveles de correlación electrónica
Los niveles de correlación electrónica con los que normalmente nos enfrentamos al trabajar sistemas, tales como sólidos (se aclara que estos niveles de correlación no solo afectan a sólidos sino también a moléculas y átomos) son:
— Los sistemas débilmente correlacionados se dan para los que poseen orbitales s y p y que normalmente no tienen gran repercusión en las propiedades de los sistemas.
— Los sistemas medianamente correlacionados se presentan en sistemas con electrones d, aunque pudieran llegar a ser fuertemente correlaciones, además aquí se inicia un mayor peso de dicho efecto en las propiedades de los sistemas.
— Los sistemas fuertemente correlacionados se presentan principalmente en compuestos que contienen iones de lantánidos y actínidos, es decir, sistemas con electrones 4f y 5f como lo son aquellos que contienen Cerio, Uranio o Neptunio como constituyentes (Fig. 1 y Tabla 2).

Pero ¿qué es la correlación electrónica (CE) y correlación electrónica fuerte (CEF)?
— Sabemos que las posiciones y movimientos de las partículas hacen que una molécula o material sean correlacionados, debido a que dichas partículas interactúan con unas y ejercen fuerzas sobre otras conforme ellas se mueven.
De esto podemos concluir que la correlación electrónica es producida por el movimiento de las partículas, sólo debemos tener en cuenta que dichas interacciones son cuasi-instantáneas (siendo el límite la velocidad de la luz). Por otro lado, tenemos que
— El concepto correlacionado fuertemente se usa para describir condiciones cuando la repulsión coulombiana entre electrones inhibe fuertemente su movimiento.
Esta inhibición de movimiento (es decir, su movimiento esta restringido por la forma misma de los orbitales f, por eso se les conoce como electrones localizados) es la que hace que no tengamos el comportamiento adecuado de los materiales que contienen lantánidos y actínidos, principalmente.
Métodos post-DFT para el tratamiento de la CEF
Las aproximaciones que trabajan correctamente para sistemas altamente correlacionados en la teoría de funcionales de la densidad, los podemos clasificar como métodos post-DFT. En la Tabla 1 damos a conocer en una forma general la estructura de algunas aproximaciones.
Dichas metodologías se basan principalmente en hacer modificaciones a la aproximación de la densidad local (LDA), por estar construida bajo la teoría del campo medio, de ahí la facilidad de modificarla y tomar en cuenta el parámetro de correlación U (desde el punto de vista de la Química Cuántica se puede interpretar dicho parámetro como la energía que necesita el electrón para pasar del nivel de energía HOMO al LUMO) e intercambio J (provenientes del principio de antisimetría o del principio de exclusión de Pauli)[15]. Las diferentes metodologías que existen son basadas prácticamente en la teoría DFT (excepto la aproximación GW), e implementadas en las aproximaciones LMTO-ASA[16] (Linear Muffin-tin Obitals-Atomic Sphere Approximation) y FP-LMTO[17] (Full Potential-Linear Muffin-tin Obitals) por la facilidad que se tiene para implementar dicha correlación fuerte (parámetro U proveniente del modelo de Hubbard, que es la introducción de Hamiltonianos modelos que tratan dicho efecto).


La aproximación LMTO-ASA tiene la a) celda unitaria dividida en esferas Muffin-Tin traslapadas (con un traslape máximo de 15 % para que se considere un cálculo aceptable, pero para el caso de estructuras abiertas (orbitales atómicos semillenos) las esferas vacías describen los potenciales de repulsión en el intersticio) y el potencial de simetría esférico dentro de las esferas, además de basarse en b) la condición ASA (Atomic Sphere Approximation, aproximación esférica sin zona de electrones libres en la estructura Muffin-Tin, es decir, se colocan esferas centradas en los átomos dentro de la celda unitaria para tener el volumen total de dicha celda) para determinar el volumen correcto de la celda.
En la segunda metodología la cual es una implementación autoconsistente de las ecuaciones de Kohn-Sham (provenientes de la ecuación de Bloch para sólidos). Dicho método expande las funciones de onda de un electrón en términos de orbitales Muffin-tin [17]. Además, este método usa una forma completamente general para el potencial y la densidad el cual el espacio es dividido en a) esferas Muffin-tin no-traslapadas centradas en sitios atómicos, b) y una región intersticial (aquí el potencial es expandido como una combinación lineal de funciones Hankel), en lugar de usar la aproximación ASA.
Efectos de la CEF en materiales sólidos
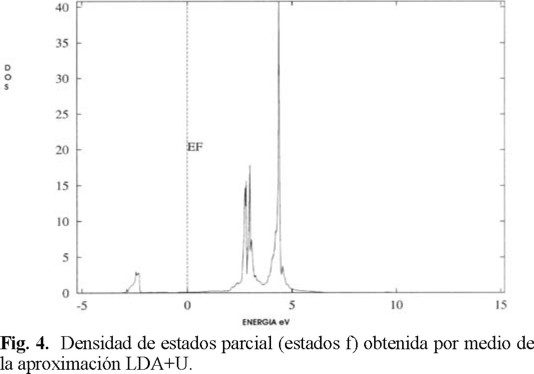
El efecto que puede tener la CEF puede ser visualizado en la densidad de estados (DOS) principalmente. Consideremos el siguiente ejemplo: el Cerio (estructura cúbica con grupo espacial 225), si observamos la gráfica de la densidad de estados al estudiarla con LDA nos predice el comportamiento de metálico (Fig. 2), dado que tenemos bandas ligeramente desplazadas del nivel de Fermi. Al estudiarla con la aproximación LDA+U (Local Density Approximation + Hubbard correction term, figura 3) [18] (por ejemplo) se predice aún el comportamiento metálico, pero se observa como se abren la banda central (formándose un pseudogap de 6 eV aproximadamente) que ante teníamos en un cierto valor U [22], así con esto tenemos estados α por debajo del nivel de Fermi y estados ß por encima del nivel de Fermi. Además la mayor contribución a la densidad estado total se tiene por parte de los electrones f (Fig. 4).


En la DOS se tiene comúnmente el comportamiento metálico al tener bandas desplazadas ligeramente del nivel de Fermi (al no tener gap de energía podemos definirla como sistema metálico), esto también se puede observar en la EB (Estructura de Bandas) en la cual tendremos bandas casi horizontales, dándonos el comportamiento de fermión pesado, característica de los sistemas altamente correlacionados.
Dicha CEF puedo provocar por ejemplo: la transición Mott (transición metal-aislante), el efecto Kondo[23] (Fig. 5, mecanismo que se presenta en metales cuando se tiene una impureza magnética, en nuestro caso el Cerio y Uranio son las impurezas), etc.

Conclusión
Con esta contribución tratamos de esclarecer el concepto de correlación electrónica fuerte (CEF), dicha interacción restringe el movimiento de los electrones localizados por la forma misma de los orbitales f (o d), esto queda corroborado con los cálculos realizados de la densidad de estados total y parcial para el ion cerio tanto a nivel DFT convencional como lo es la aproximación LMTO-ASA, así como por medio de las correcciones hechas a dicha aproximación llamada LDA+U. También, tenemos ciertos tipos de materiales que son afectados por esta CEF como los superconductores a alta Tc y los sistema de fermiones pesados, siendo los sistemas actualmente los de mayor interés científico por presentar diversidad de propiedades físicas y químicas.
Lo que es importante señalar que no solo afecta a sólidos (que es el enfoque que se le dio a dicho artículo) sino también a sistemas de interés químico como metales orgánicos (conocidos como metales mesoscópicos)[24], y compuestos basados en carbonos como nanotubos de carbón (dependiente de cómo sea enrollado presentan propiedades de semiconductor o metal [25], Fig. 7), fulerenos[26] (Fig. 8), etc., que presentan efectos de la alta correlación electrónica.



Agradecimientos
Se agradece al Centro de Cómputo del Instituto de Física "Luis Rivera Terrazas" de la Benemérita Universidad Autonoma de Puebla las facilidades otorgadas para la realización de esta trabajo. Trabajo apoyado por CONACyT, México, proyecto No. 32213-E y por la Vicerrectoría de Investigación y Estudios de Posgrado (VIEP) de la Benemérita Universidad Autónoma de Puebla, proyecto No. II-101I02.
Referencias
1. Chigo-Anota, E.; Rivas Silva, J. F. Rev. La Ciencia y El Hombre, Universidad Veracruzana, 2003. [ Links ]
2. Chigo-Anota, E. Rev. Avance Tecnológico y Sociedad, 2002, 24, 33-37, Universidad Tecnológica de Puebla. [ Links ]
3. Chigo-Anota, E. Tesis de Maestría en Ciencias, Instituto de Física "LRT" de la Benemérita Universidad Autónoma de Puebla, 2000; [ Links ] Slater, J. C. Phys. Rev. 1951, 81, 385-390. [ Links ]
4. Kohn, W.; Becke A. D.; Parr, R. G. J. Phys. Chem. 1996, 100, 12974-12980; [ Links ] Capelle, K. Preprints en el archivo de internet cond-mat/0211443 V1, Los Alamos Laboratory, Noviembre, 2002. [ Links ]
5. Frisch, Æ.; Frisch, M. J. User´s Reference (Gaussian, Inc.), 1998; [ Links ] Foresman, B. J.; Frisch, Æ. Exploring Chemistry with Electronic Structure Methods, Ed., 1996. [ Links ]
6. Dovesi, R.; Saunders, V. R.; Roetti, C. CRYSTAL92, An ab-initio Hartree-Fock LCAO program for periodic systems, 1992; [ Links ] Pisani, C.; Dovesi, R.; Roetti, C. Hartree-Fock Ab Initio Treatment of Crystalline Systems, Springer Verlag, 1988. [ Links ]
7. Cerius2 versión 4.2 MatSci, Manual de CASTEP y ESOCS, Molecular Simulations Inc. 2000. [ Links ]
8. Andersen, O. K. Phys. Rev. B 1975, 12, 3060-3083; [ Links ] Skriver, H. L. The LMTO method, Springer-Verlag Berlín, 1984. [ Links ]
9. Sing, D. J. Planewaves, Pseudopotentials and the LAPW Method, Kluwer Academic, 1994. [ Links ]
10. Fulde, P. J. Phys. F: Met. Phys. 1988, 18, 601-639; [ Links ] Stewart, G. R. Rev. Mod. Phys. 1984, 56, 755-787. [ Links ]
11. Yamamoto, H.; Sawa, H.; Ishikawa, M. Phys. Lett. A 1994, 196, 83-86; [ Links ]Yamamoto, H.; Ishikawa, M.; Hasegawa, K.; Sakurai, J. Phys. Rev. B 1995, 52, 10136-10141; [ Links ] Chigo Anota, E.; Flores Riveros, A.; Rivas Silva, J. F.; Bautista Hernández, A.; TNFCM-V (Quinto Taller Nacional de Física y de Ciencias de Materiales para estudiantes de postgrado), 2002, 21, 131-136; [ Links ] Chigo Anota, E.; Rivas-Silva J. F.; Bautista Hernández, A.; Flores Riveros, Superficies y Vacío 2003, 16, 17-21. [ Links ]
12. Chigo-Anota, E.; Bautista Hernández, A.; Rivas-Silva, J. F. Rev. Méx. Fís. 2003 (enviado). [ Links ]
13. Chigo Anota, E.; Rivas Silva, J. F. Superficies y Vacío 2002, 14, 7-11. [ Links ]
14. Sandratskii, L.M.; Kübler, J.; Zahn, P.; Mertig, I. Phys. Rev. B 1994, 50, 15834-15842; [ Links ] Takahashi, T.; Sato, N.; Yokoya, T.; Chainani, A. Morimoto, T.; Komatsubara, T. J. Phys. Soc. Japan 1996, 65, 156-159; [ Links ] Metoki, N.; Haga, Y.; Koike, Y.; Onuki, Y. Phys. Rev. Lett. 1998, 80, 5417-5420. [ Links ]
15. En la estructura Hartree-Fock existen tres interpretaciones, la primera, su cantidad total es una carga electrónica si ui es uno de los orbitales de espín ocupado pero es cero si ui es un orbital de espín excitado. Segunda, propiedad de la densidad de carga de intercambio es que consiste de una carga electrónica que tiene el mismo espín como el orbital de espín ui. Tercero, cuando estas anteriores condiciones son iguales, la densidad de carga de intercambio se convierte en una sumatoria de orbitales o es la densidad total de todos los electrones del mismo espín.
16. Andersen, O. K.; Jepsen, O. Phys. Rev. Lett. 1984, 53, 2571-2574. [ Links ]
17. Methfessel, M.; Schilfgaarde, M.V. Phys. Rev. B 1993, 48, 4937-4940; [ Links ] Methfessel, M.; Phys. Rev. B 1988, 38, 1537-1540; [ Links ] Methfessel, M.; Rodríguez, C.O.; Andersen, O.K. Phys. Rev. B 1989, 40, 2009-2012; [ Links ] Qui, M.; Jiang, M.; Zhao, Y.J.; Cao, P.L.; J. Chem. Phys. 1999, 110, 10738-10745. [ Links ]
18. Anisimov, V. I.; Solovyev, I. V.; Korotin, M.A.; Czyzyk, M.T.; Sawatzky, G.A. Phys. Rev. B 1993, 48, 16929-16934; [ Links ] Anisimov, V. I.; Aryatesiawan, F.; Lichtenstein, A. I. J. Phys.: Condens. Matter 1997, 9, 767-808. [ Links ]
19. Lundin U.; Eriksson, O. Int. J. Quantum Chem. 2001, 81, 247. [ Links ]
20. Aryatesiawan F.; Gunnarsson, O. Preprints en el archivo de internet cond.-mat/971203 v1. Los Alamos Laboratory, diciembre 1997. [ Links ]
21. Lichtenstein, I.; Katsnelson, M. I. Phys. Rev. B 1998, 57, 6884-6895. [ Links ]
22. valor obtenido y no reportado por Rivas Silva J. F.; Anisimov V. I.; Gunnarsson, O. Phys. Rev. B 1991, 43, 7570-7574; [ Links ] Min, B. E.; Jansen, H. J. F.; Oguchi, T.; Freeman, A. J. Phys. Rev. B 1986, 33, 8005-8015. [ Links ]
23. Kouwenhoven, L.; Glazman, L. Physics World 2001, 14, 33-38. [ Links ]
24. http://www.zipperlig.de/News/orgmetal.en.html; Ishikawa, A.; Matsunaga, N.; Nomura, K.; Sasaki, T.; Nakamura, T.; Takahashi, T.; Saito, G. Phys. Rev. B. 2003, 67, 212404-212107. [ Links ]
25. http://www.phys.psu.edu/~crespi/research/carbon.ld/public; propiedades como superconductividad también se presentan en nanotubos de carbón consultar: Zhao, G. M. preprints en el archivo de internet cond-mat/0307770 v3. Los Alamos Laboratory, agosto 2003. [ Links ]
26. Andreoni, W. Annu. Rev. Phys. Chem. 1998, 49, 405-439; [ Links ] Forró, L.; Mihaly, L. Rep. Prog. Phys. 2001, 64, 649-699; [ Links ] Koch, E.; Gunnarsson, O.; Wehrli, S.; Sigrist, M. preprints en el archivo de internet cond-mat/0305274 v1. Los Alamos Laboratory, Mayo 2003. [ Links ]

