










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de la Sociedad Química de México
Print version ISSN 0583-7693
Rev. Soc. Quím. Méx vol.47 n.3 Ciudad de México Jul./Sep. 2003
Investigación
Assignment of the 1H and 13C NMR spectra of N2,N6-dibenzoyl-N2,N9-bis(2',3'-di-O-benzoyl-(α)-L-threofuranosyl)-2,6-diaminopurine
Guillermo Delgado1* and Ramanarayanan Krishnamurthy2*
1 Instituto de Química de la Universidad Nacional Autónoma de México. Circuito Exterior, Ciudad Universitaria. Coyoacán 04510, México, D. F. E-mail: delgado@servidor.unam.mx
2 Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, U.S.A. E-mail: rkrishna@scripps.edu
Recibido el 12 de mayo del 2003
Aceptado el 18 de agosto del 2003
Abstract
The complete 1H- and 13C NMR-chemical shifts assignments of the title 2,6-diaminopurine nucleoside are described. Spectroscopic analysis confirmed the R- configuration at the anomeric carbons and that both the threofuranoses exist in a conformational equilibrium at room temperature.
Keywords: 1H and 13C NMR spectroscopy; 2,6-diaminopurine; nucleosidation; (α)-L-threofuranose; nucleosides, N2,N6-dibenzoyl-N2,N9-bis(2',3'-di-O-benzoyl-(α)-L-threofuranosyl)-2,6-diaminopurine; TNA.
Resumen
Se describen las asignaciones de RMN 1H y 13C del nucleósido derivado de 2,6-diaminopurina nombrado en el título del trabajo. El análisis espectroscópico confirmó la configuración R- de los carbonos anoméricos, y ambas treofuranosas existen en un equilibrio conformacional a temperatura ambiente.
Palabras clave: Espectroscopía de RMN 1H y 13C; 2,6-diaminopurina; nucleosidación; (α)-L-treofuranosa; nucleósidos, N2,N6-dibenzoil-N2,N9-bis(2',3'-di-O-benzoil-(α)-L-treofuranosil)-2,6-diaminopurina; ATN.
Introduction
The systematic study of the base-pairing properties of various sugar-modified-backbone analogs of nucleic acids, carried out by Eschenmoser and co-workers [1], has led to the discovery of α-L-threofuranosyl-(3' → 2')-oligonucleotides ("TNA") as an informational pairing system in an antiparallel strand orientation that is also capable of cross-pairing with complementary sequences of RNA and DNA [2]. TNA is derived from threose-sugar containing only four carbon atoms and is one of the simplest potentially natural nucleic acid alternatives investigated, in the context of a chemical etiology of nucleic acid structure. In this context it was further shown, by Eschenmoser and coworkers [3], that replacing adenine with 2,6-diaminopurine nucleobases in TNA-sequences results in enhanced thermal- and thermodynamic-stability of TNA/TNA-, TNA/RNA- and TNA/DNA-duplexes and accelerates template directed ligation of TNA ligands. The 2,6-diaminopurine-threose-nucleoside 1a, which is the initial building block required for the automated solid-support synthesis of TNA-sequences, was synthesized by nucleosidation of the peracylated threose 2 with N2,N6-dienzoyl-2,6,-diaminopurine 3 (D(Bz)2) under the standard Hilbert-Johnson-Vorbrüggen conditions (Scheme 1) [4]. Thus, an initial exploratory reaction of 2 with silylated 3 in the presence of 1.2 eq. of SnCl4 as Lewis acid [5,6] afforded, apart from the epimeric mixture of N9-nucleosides 1a (24 %) and 1b (8 %), a compound 4 which is formed in minor amounts (6 %). Compound 4 turned out to be, unexpectedly, a N2,N9-bisnucleoside derivative of 2,6-diaminopurine. Here, we report on the structure determination of 4 and its 1H- and 13C-NMR assignments with the aid of 2D-NMR measurements (1H-1H COSY, DEPT, HMQC, HMBC and NOESY).
Results and Discussion
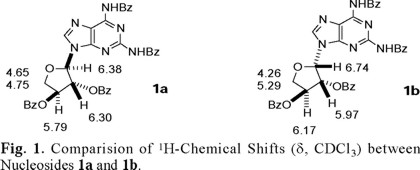
Structure of compound 1a was established on the basis of multidimensional NMR spectroscopic data and by an X-ray structure analysis of the 2',3'-diol derivative obtained by the selective hydrolysis the 2',3'-benzoate groups of 1a [5]. Compounds 1a and 1b exhibited the same MS-data corresponding to the formula C37H28N6O7; they also displayed the same connectivity in the HMBC experiments, and the C-1'-N-9 linkage was determined by the presence of H-1'-C-4 crosspeaks, thus ruling out the possibility that 1b could be the regioisomeric N7-nucleoside. Comparison of the 1H-chemical shifts between 1a and 1b (Fig. 1) coupled with MS-data further confirmed the epimeric relationship between the two nucleosides. A couple of noteworthy differences in the spectroscopical behaviour between the two nucleosides are: (a) NOESY correlation was observed between H-8 and H-1' for nucleoside 1b which was absent in nucleoside 1a and (b) the coupling constant between H-1' and H-2' for nucleoside 1a (J1',2') = 2 Hz, while for nucleoside 1b (J1',2') = 5 Hz.
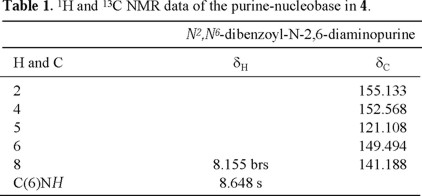
Tables 1-4 (2, 3) list the 1H- and 13C-NMR data for compound 4. ESI-MS for compound 4 indicated a molecular formula of C55H42N6O12. This molecular formula agreed with the structure of a N2,N6-dibenzoyl-2,6-diaminopurine (C5H2N6(C7 H5O)2=C19H12N6O2) linked to two dibenzoylated threoses (2(C18H15O5)=C36H30O10). The 13C-NMR spectrum of 4 showed 43 signals, 8 of which correspond to the two threoses (rings A and B, see Fig. 2), 5 to the purine nucleus, 6 to the carbonyl groups, and the remaining 24 signals to the corresponding non-equivalent carbons of the six benzene rings. The individual spin systems were discerned from the HMQC and HMBC experiments and the 1H- and 13C-NMR assignments of the N2,N6-dibenzoyl-2,6-diaminopurine fragment are shown in Table 1. The amide proton (δH 8.648, exchanges with D2O) showed HMBC correlation with C-5 (δC 121.108), which in turn correlated with H-8 (δH 8.155). This last proton showed a cross-peak with the secondary carbon located at δC 141.188 in the HMQC experiment.
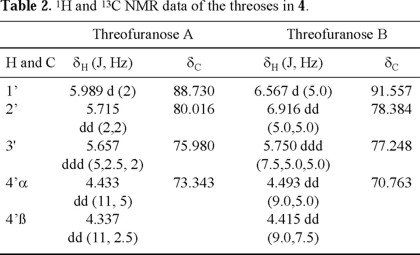
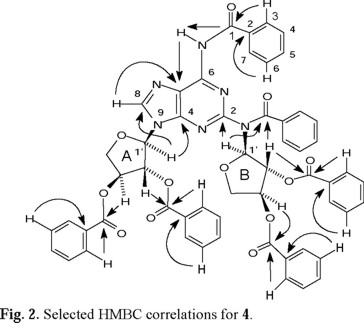
1H-1H COSY NMR experiments clearly indicated the presence of two sets (A and B) of the sugar protons (see Table 2). The signals at δC 88.730 (set A) and δC 91.557 (set B, determined as the anomeric carbons, due to the observed HMBC crosspeaks, vide infra) correlated with the hydrogens at δH 5.989 and 6.567, respectively, in the HMQC spectra, locating the anomeric hydrogens (Table 2). The connectivities of anomeric carbons of the threoses to N-9 and to the nitrogen at C-2 were established by the observed HMBC correlation between H-1'A and C-4 (δC 152.568) and between H-1'B and C-2 (δC 155.133). Selected HMBC correlations that allowed the full spectral assignments are shown in Fig. 2.
The stereochemistry at the anomeric carbons was determined by the presence of cross-peaks in the NOESY spectrum between H-1' and H-3' for both threoses (Fig. 3), determining their cis-relationship. This existence of NOESY between H-1' and H-3' could be possible only when the nucleoside is an α-anomer. Therefore the configuration at C-1'A and C-1'B is assigned to be R.
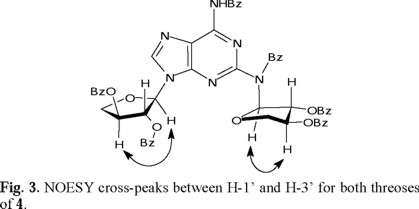
This NOESY correlation is weak for the ring A and strong for the ring B at room temperature, suggesting unequal conformational populations for each threose. The observed 1H-1H coupling constants between H-1' and H-2' are also different (J1',2' = 2.0 Hz for ring A, and J1',2' = 5.0 Hz for ring B, see Table 2), suggesting that the average conformations for both threoses are different. This is in agreement with the NOESY interactions between H-1' and H-2', between H-2' and H-3', and between H-1' and H-3', which were observed to be different for both threoses. These observations underline the limitations in assigning the relative configuration at C(1') considering only the magnitude of the J1',2' coupling constants. The schematic representations of the two principal puckering modes for the threose ring, namely, C(2')-endo (S) and the C(3')-endo (N) that explain the observed NOESY correlations for both threoses of 4, are shown in Fig. 4.
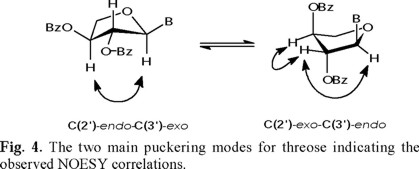
This conformational interconversion [C(2')-endo (S)⇌ C(3')-endo (N)] is well documented for the ribofuranosyl series in the literature, and the barrier of the conformational equilibrium is relatively low [7,8]. The average activation energy has been estimated to be 4.7 ± 0.5 kcal/mole for purine ribonucleosides.
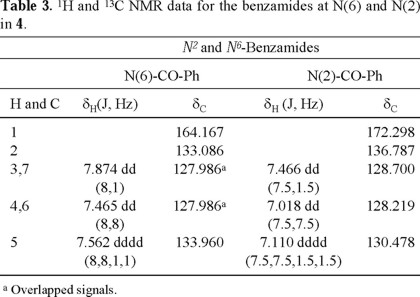
The 1H- and 13C-NMR for the two benzamides and the four benzoates are shown in Tables 3 and 4, respectively, and were assigned by identifying the HMBC correlations for each carbonyl group with the corresponding o,m-hydrogens (H-3, H-7 and H-4, H-6), and in the case of the benzoates, also identifying the interactions with the sugar hydrogens. The carbonyl of the benzamide at C-6 (δC 164.167) showed a cross-peak with the amide hydrogen, while the benzamide at C-2 (δC 172.298) showed a cross-peak with the anomeric hydrogen H-1'B, as shown in Fig. 2. Some assignments can be interchanged and several signals appeared overlapped, as indicated in Tables 3 and 4.
The observed downfield chemical shift of H-C(2') in threose B (H-2'B, δ 6.92) of 4 deserves a comment. One possible explanation is the proximity of this hydrogen to a heteroatom; and in principle, there are at least 3 conformational possibilities, which would result in such proximity. These are (a) the carbonyl group of the benzamide linked to C(2) close to H-2'B (conformer A); (b) H-2'B close to N(3) (conformer B), and free rotation along the sigma C(2)-N bond generates another arrangement in which (c) H-2'B close to N(1) (conformer C). These conformational possibilities are shown in Fig. 5.
Simple hand held molecular model considerations reveal that it is not possible with any degree of certainty to single out one conformer out of the three possibilities considered here; though there seems to be a preference for the sterically less hindered conformer C. However, the observed data correspond to an average structure rather than a single conformer, since the NOESY interactions show evidence of the conformational mobility of 4 at room temperature.
Experimental
N2,N6-dibenzoyl-N9-(2',3'-di-O-benzoyl-(α)-L-threofuranosyl)-2,6-diaminopurine (1a), N2,N6-dibenzoyl-N9-(2',3'-di-O-benzoyl-(β)-L-threofuranosyl)-2,6-diaminopurine (1b), N2,N6-dibenzoyl-N2,N9-bis(2',3'-di-O-benzoyl-(α)-L-threofuranosyl)-2,6-diaminopurine (4). In a dried three necked round bottom flask equipped with a condenser and a septum was placed 1.1 g (2.96 mmol, 1.02 eq.) of N2,N6-dibenzoyl-2,6-diaminopurine 3 (previously dried in the high vacuum at 70 °C, using the Büchi gun) and solution of 1.1 g (2.90 mmol, 1.0 eq.) of peracylated sugar 2 in 9 mL of MeCN was added by a syringe. Additional 3 mL of MeCN was used to rinse the flask. The reaction mixture was heated to 80 °C with stirring under Ar. After 10 min, 2.78 mL (11.2 mmol, 3.86 eq.) of bis(trimethylsilyl)acetamide (BSA) was added, obtaining a clear solution. After 30 min, 415 µL (3.5 mmol, 1.22 eq.) of SnCl4 was added and the heating was maintained for 2 h. The solution turned dark brown. The reaction mixture was cooled to room temp., poured to a stirred solution of EtOAc and satd. aq. NaHCO3 (150 mL each). The phases were separated, and the aqueous phase was extracted two times with additional portions of EtOAc. The organic phases were combined, washed with NaHCO3, brine, and dried (MgSO4). The organic residue was concentrated to dryness to afford a residue (1.4 g), which was separated by column chromatography (60 g of silica gel packed with hexane-EtOAc (55:45), and increasing the polarity to 1:1, 3:2, and 7:3). From the fractions eluted with hexane-EtOAc (1:1) was obtained a residue (320 mg), and part of this (81 mg) was rechromatographed with silica (7 g) suspended in hexane/EtOAc (85:15) and increasing the proportion of EtOAc to 75:25, 70:30, 65:35, 60:40, 1:1. This procedure afforded 43 mg of 4 (6 %) as a yellow solid. From the fractions eluted with hexane/EtOAc (7:3) of the original column chromatography, a residue (717 mg) was obtained, which was purified by column chromatography (∅ 2.5 × 40 cm, 100 g of silica gel) and a mixture of hexane/benzene/EtOAc/CH2Cl2 (10:10:2.5:2.5) with gradually increasing polarity by the addition of methanol (0.1 → 0.4 %) to this mixture) to afford 470 mg of 1a (24 %) and 150 mg of 1b (8 %).
N2,N6-dibenzoyl-N9-(2',3'-di-O-benzoyl-(α)-L-threofuranosyl)-2,6-diaminopurine (1a). For complete characterization see reference 5.
N2,N6-dibenzoyl-N9-(2',3'-di-O-benzoyl-(β)-L-threofuranosyl)-2,6-diaminopurine (1b). Mp. 116-118°C, TLC (benzene / EtOAc / CH2Cl2 / MeOH 8:1:1:0.2): Rf 0.45. 1H NMR (500 MHz, CDCl3): 4.26 (dd, J = 9.5, 2.5, H-C(4'a)); 5.19 (dd, J = 9.5, 5.0, H-C(4'b)); 5.97 (dd, J = 5.0, 2.5, H-C(2')); 6.17 (br s, H-C(3')); 7.2-8.2 (m, 20 arom. H); 8.06 (s, H-C(8)); 9.38 and 9.53 (br s, HNCO). 13C NMR (125 MHz, CDCl3): 72.53 (t, C(4')); 74.67 (d, C(2')); 77.17 (d, C(3'); 84.82 (d, C(1')); 119.58 (s, C(5)); 141.64 (d, C(8)); 149.75 (s, C(6)); 152.34 (s, C(4)); 152.68 (s, C(2)); 165.19 (s, 2CO benzamido); 165.54, 165.81 (2s, CO). MALDI-FTMS, m/z: 691.1892 (100, [M +Na]+).
N2,N6-dibenzoyl-N2,N9-bis(2',3'-di-O-benzoyl-(α)-L-threofuranosyl)-2,6-diaminopurine (4). Mp. 122-124 °C, TLC (hexane / EtOAc 1:1): Rf 0.35. 1H NMR and 13C NMR: see Tables 1-4 (2, 3). ESI-MS (pos.), m/z: 979 (100, [M + H]+); ESI-MS (neg.), m/z: 977 (38, [M − H]−).
Spectra. All the experiments were performed at room temperature on a Varian Unity Plus 500 spectrometer operating at 499.89 MHz for 1H and 125.71 MHz for 13C. TMS was used as an internal reference for the spectra, and CDCl3 as solvent. The COSY spectra were acquired with a spectral width of 4827 Hz in f2 and f1, with 16 transients and 128 t1 increments. The pulse programs of COSY, HMQC, HMBC and NOESY experiments were taken from the Varian software library and were obtained using an indirect detection probe. The HMQC and HMBC (9 Hz) were collected using a spectral width of 4827 Hz for f2 (1H) and 21751 Hz for f1 (13C) and 26109 Hz for HMBC. With 1024 data points, a relaxation delay of 1s using 32 transients (HMQC) and 128 transients (HMBC) both with 256 increments. Data were processed with linear prediction to 2048 points in the indirect detected frequency.
Acknowledgements
The support of the Skaggs Research Foundation (TSRI) and the Universidad Nacional Autónoma de México (Dirección General de Asuntos del Personal Académico) is gratefully acknowledged. This work was made possible by the support of Professor Albert Eschenmoser and was carried out in his laboratory in the context of studies on the chemistry of TNA.
References
1. Eschenmoser, A. Science 1999, 284, 2118-2124 and references cited therein. [ Links ]
2. Schöning K.-U.; Scholz, P.; Guntha, S.; Wu, X.; Krishnamurthy, R.; Eschenmoser, A. Science 2000, 290, 1347-1351. [ Links ]
3. Wu, X.; Delgado, G.; Krishnamurthy, R.; Eschenmoser, A. Org. Lett. 2002, 4, 1283-1286. [ Links ]
4. (a) Hilbert, G. E.; Johnson, T. B. J. Am. Chem. Soc. 1930; 52, 4489-4491; [ Links ] (b) Vorbrüggen, H.; Bennua, B. Chem. Ber. 1981, 114, 1279-1286. [ Links ]
5. Schöning, K.U.; Scholz, P.; Wu, X.; Guntha, S.; Delgado, G.; Krishnamurthy, R.; Eschenmoser, A. Helv. Chim. Acta 2002, 85, 4111-4153. [ Links ]
6. It has been noted that the amount of SnCl4 plays an important role in the N7/N9 selectivity in nucleosidation reactions. Parel, S.; Leumann, C. H. Helv. Chim. Acta 2000, 83, 2514-2526. [ Links ]
7. Saenger W. Principles of Nucleic Acid Structure. Springer-Verlag. 1988. [ Links ]
8. Blackburn; G. M.; Gait, M. J. (Eds.) Nucleic Acids in Chemistry and Biology. IRL Press at Oxford University, 1990. [ Links ]

