










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
 Cited by SciELO
Cited by SciELO Related links
 Similars in
SciELO
Similars in
SciELO Share

 Permalink
PermalinkRevista de la Sociedad Química de México
Print version ISSN 0583-7693
Rev. Soc. Quím. Méx vol.47 n.2 Ciudad de México Apr./Jun. 2003
Investigación
New Eremophilanoids from the Roots of Psacalium radulifolium. Hypoglycemic, Antihyperglycemic and Anti-Oxidant Evaluations
María Luisa Garduño-Ramírez1 and Guillermo Delgado2,*
1 Centro de Investigaciones Químicas de la Universidad Autónoma del Estado de Morelos, Avenida Universidad 1001, Chamilpa 62210, Cuernavaca, Morelos, México.
2 Instituto de Química de la Universidad Nacional Autónoma de México, Circuito Exterior, Ciudad Universitaria, Coyoacán 04510, México, D.F. E-mail: delgado@servidor.unam.mx
Recibido el 21 de abril del 2003.
Aceptado el 23 de junio del 2003.
Dedicated to Dr. Alfonso Romo de Vivar.
Abstract
The investigation of the chemical constituents from the roots of Psacalium radulifolium (Compositae), a member of the matarique complex of medicinal plants, resulted in the isolation of four additional new eremophilanoids: radulifolin D, radulifolin E (ketodecompostin), radulifolin F (3β-hydroxycacalone-3-O-β-D-glucopyranoside) and epi-radulifolin F (3β-hydroxy-6-epi-cacalone-3-O-β-D-glucopyranoside), together with the known compounds maturinone, acetylmaturine, dimaturine, triacontanol, hydroxycacalolide, epi-hydroxycacalolide, β-sitosteryl-3-O-β-D-glucopyranoside, β-D-glucopyranose and saccharose. The methanol extract from the roots of this species displayed hypoglycemic activity, but cacalol, cacalone, epi-cacalone, O-methyl-1,2-dehydrocacalol and decompostin did not exhibit activity. The antihyperglycemic evaluation of the extract demontrated that it was inactive. Some isolated compounds were also tested for antioxidant activity, and cacalol was found to be active.
Keywords: Psacalium radulifolium, Compositae, matarique, radulifolin D, radulifolin E, radulifolin F, epi-radulifolin F, eremophilanoids, hypoglycemic activity, antihyperglycemic activity, anti-oxidant activity.
Resumen
La investigación de los constituyentes químicos de las raíces de Psacalium radulifolium (Compositae), una especie perteneciente al complejo matarique de plantas medicinales, resultó en el aislamiento de cuatro nuevos eremofilanoides: radulifolina D, radulifolin E (cetodecompostina), radulifolina F (3-O-β-D-glucopiranósido de 3β-hidroxicacalona) y epi-radulifolina F (3-O-β-D-glucopiranósido de 3β-hidroxi-6-epi-cacalona), junto con las substancias conocidas maturinona, acetil maturina, dimaturina, triacontanol, hidroxicacalólida, epi-hidroxicacalólida y 3-O-β-D-glucopiranósido de β-sitosterilo, β-D-glucopiranosa y sacarosa. El extracto metanólico de las raices de esta especie mostró actividad hipoglucémica, pero cacalol, cacalona, epi-cacalona, el éter metílico de 1,2-deshidrocacalol y la decompostina no mostraron actividad. La evaluación antihiperglucémica del extracto demostró su inactividad. La actividad antioxidante fue ensayada para algunas substancias, y se encontró que el cacalol es activo.
Palabras clave: Psacalium radulifolium, Compositae, matarique, radulifolina D, radulifolina E, radulifolina F, epi-radulifolina F, eremofilanoides, actividad hipoglicémica, actividad antihiperglicémica, actividad antioxidante.
Matarique is the common name for a group of plants used in Mexican traditional medicine for the treatment of diabetes, kidney pains, infections, and general body pains, among other ailments [1-4]. This group includes Psacalium decompositum, P. palmeri, P. peltatum, P. sinuatum, and A. thurberi. Psacalium belongs to the Tussilaginoid genera of the Senecioneae (Compositae), and includes ca. 40 species which are located chiefly in Mexico [5]. The structures and chemistry of the secondary metabolites isolated from P. decompositum (syn: Cacalia decomposita) have been the subject of several investigations [6], and the synthesis of the main constituents, cacalol (1), cacalone (2) and structural analogs have been achieved [7]. Cacalol (1) has been found as the bioactive constituent in antimicrobial [8], antioxidant [9], allelopatic and phytopathogenic assays [10]. The hypoglycemic activity of extracts of P. decompositum and P. peltatum in mice have been evaluated [11,12], and the antihyperglycemic activity of aqueous extracts and some constituents from P. decompositum using diabetic mice have been determined [13].
P. radulifolium is considered a substitute for the preferred P. decompositum in the matarique complex of medicinal plants, and previous examination of the less polar constituents of the roots of this species allowed the isolation of 1, 2, epi-cacalone (3), radulifolin A (4), epi-radulifolin A (5), radulifolin B (6), radulifolin C (7), O-methyl-1,2-dehydrocacalol (8), adenostin A (9), decompostin (10) and neoadenostylone (11), from which 1 displayed major antimicrobial activity [14]. Here we report the hypoglycemic and antihyperglycemic evaluations of the methanol extract of the roots of P. radulifolium, and the isolation of the polar constituents, which resulted in the characterization of four new metabolites: radulifolin D (12), radulifolin E (ketodecompostin, 13), radulifolin F (3β-hydroxycacalone-3-O-β-D-glucopyranoside, 14) and epi-radulifolin F (3β-hydroxy-6-epi-cacalone-3-O-β-D-glucopyranoside, 15), together with the known compounds maturinone (16), acetylmaturine (17), triacontanol, dimaturine (18), hydroxycacalolide (19), epi-hydroxycacalolide (20), β-sitosteryl-β-D-glucopyranoside, β-D-glucopyranose and saccharose. The antioxidant activity of some isolated compounds is also reported.
Results and discussion
Compound (12) was isolated as a yellow solid, and its HREIMS established the molecular formula C15H14O4. UV spectrum showed bands of a conjugated ketone at lmax 336, 277, 245, and 207 nm, and the IR spectrum revealed the presence of hydroxyl group (3583 cm−1), conjugated carbonyl (1664 cm−1) and multiple carbon-carbon bonds (1585, 1463 cm−1). The 1H NMR spectrum (Table 1) showed considerable similarity with that of radulifolin C (7), a compound previously isolated from this species [14], establishing a close structural relationship. The most significant difference was the downfield shift and multiplicity of H-14 (δ 1.85), which appeared as a singlet, in comparison with the chemical shift of the same protons of radulifolin C (δ 1.43), which resonated as a doublet, indicating the presence of a hydroxyl at C-6 in 12, in agreement with the molecular formula. 13C NMR data showed the expected chemical shifts, and the assignments were corroborated by HMQC and HMBC experiments. Therefore this substance was 6-hydroxy-radulifolin C, and named radulifolin D (12). The 3-O-methyl derivative of 12 has been characterized from a chemical analysis of Cacalia hastata L. var. tanakae [15], but it was considered as an artifact due to the lack of optical activity. Radulifolin D (12) is dextrorotatory, while radulifolin C (7) is levorotatory. Considering that the twisting of the A/C rings is in the opposite direction to the pseudo-axial methyl group at C-6 (to avoid interactions with the methyls at C-13 and C-15), the hydroxyl group at C-6 of radulifolin D could be tentatively proposed with the β- configuration (12), to explain the opposite specific rotation to that observed for radulifolin C (7).
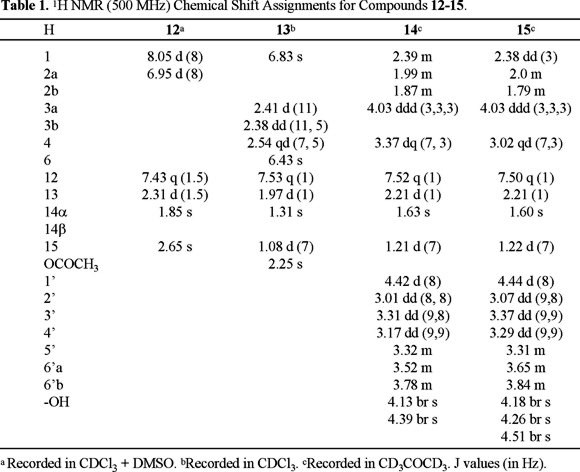
Radulifolin E (13) was isolated as a UV active solid (λmax 320, 280, 257 nm), with a molecular formula C17H18O5 established from EIMS and NMR data. The IR spectrum contained bands at 1740 and 1668 cm−1 consistent with the presence of an acetate and an α,β-unsaturated ketone, respectively. The 13C NMR spectrum (Table 2) showed 17 signals (four methyls, one methylene, four methines and eight quaternary carbons, including three carbonyls), and the chemical shifts and multiplicity observed in the 1H NMR spectrum (Table 1) could be accounted by the furanoeremophilane skeleton with an acetate at C-6 similar to that of decompostin (10). The major difference between radulifolin E (13) and decompostin was the downfield chemical shift for H-1, due to the presence of a ketone at C-2. Therefore, radulifolin E (13) was established as 2-ketodecompostin, previously obtained as a derivative of decompostin via bromination (NBS) and oxidation (AgNO3) [6d]. 1H and 13C NMR assignments (Tables 1 and 2) were confirmed by HMQC and HMBC experiments.


The FABMS, 1H and 13C NMR data for compounds 14 and 15 were consistent with the molecular formula C21H28O9. An intense IR band at ca. 3400 cm−1 for both compounds suggested the presence of several hydroxyl groups, and bands at ca. 1660 cm−1 were ascribed to α,β-unsaturated carbonyl groups. 1H and 13C NMR spectra (Tables 1 and 2) indicated the presence of a β-D-glucopyranose fragment and a cacalone aglycon. The anomeric hydrogen of 14 was observed at δ 4.42 and the analysis of the COSY spectrum determined the sequential vecinity for the carbinolic hydrogens of the β-D-glucopyranose (δ 3.78 - δ 3.01), which correlated with the corresponding signals at δ 74.78 (C-2'), 77.65 (C-3'), 71.64 (C-4'), 77.60 (C-5'), 62.97 (C-6') in the HMQC spectrum. The crosspeaks in the HMBC spectrum between H-3 (δ 4.03) and H-2' (δ 3.01) with C-1' (δ 102.71) confirmed that the glucopyranose was bound to C-3 of the modifoed eremophilane. NOESY interactions between H-1' and H-3, and between H-3 and H-4, confirmed the stereochemical assignments. Similar crosspeaks were observed for 15, and therefore, the difference between the two substances was in the aglycon fragment. The stereochemistry at C-6 was deduced by comparing the 1H NMR data of 14 and 15 with those of 2 and 3 [7d]. Specifically, the downfield shift of H-4 in 14 (δ 3.37) with respect to that of H-4 in 15 (δ 3.18) was in agreement with the α- and β- orientation of the hydroxyl groups, respectively [16].
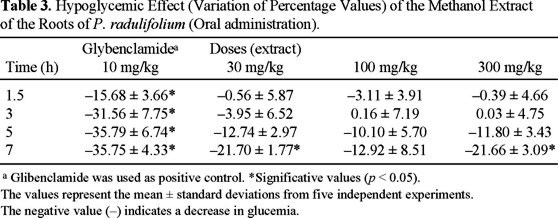
The methanol extract obtained directly from the roots of P. radulifolium was evaluated as hypoglucemic agent in normoglycemic rats, following the standard procedures [17], and the results are shown in Table 3. This residue showed significant decrease of blood glucose concentration (p < 0.05) at 7 h using several doses (30, 100 and 300 mg/kg), without returning to the basal blood glucose level. The effect of the hypoglycemic model drug is also included (glybenclamide, 10 mg/kg). Cacalol (1), The mixture of 2 + 3, O-methyl-1,2-dehydrocacalol (8), and decompostin (10) did not display significant hypoglycemic activity at doses of 3.1, 10 and 31 mg/kg.
The antihyperglycemic effect of the methanol extract was also tested at the same doses using male diabetic Wistar rats, diabetized via streptozotocin injection, following the standard procedures. The results obtained indicated that this residue did noy display significant antihyperglycemic effect in comparison with the model drug.
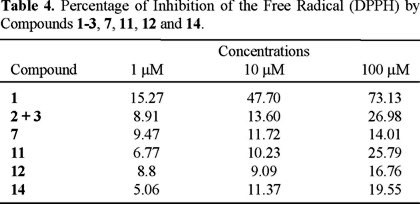
It has been proposed that the anti-oxidant activity may play a role in the antihyperglycemic and hipoglycemic activities [18], and some reactive oxygen species (superoxide anion, hydrogen peroxide and hydroxyl fee radical) are involved in the physiology of several diseases, including diabetes [19]. Therefore, the antioxidant activity of some isolated compounds (cacalol (1), the mixture of cacalone and epi cacalone (2 + 3), radulifolin C (7), neoadenostylone (11), radulifolin D (12) and radulifolin F (14)) was evaluated via the interaction with the stable free radical DPPH, following the procedures described in the experimental section. The results are included in Table 4, and they showed that only cacalol (1) exhibited significant activity, in agreement with previous reports [20].
It is interesting to note that some pure secondary metabolites did not display hypoglicemic effect, while the activity the methanolic extract of the roots of P. radulifolium was evident. Although the mechanism of action of the eremophilanes, modified eremophilanes or other natural products [21] in changing the blood glucose levels remains unknown, the described results suggest that there is some correlation with the ethnomedical use of the plant as antidiabetic agent. However, this plant, as all the plants used in traditional medicine, should not be used until safety studies are completed.
Experimental Section
General Experimental Procedures. Melting points are uncorrected. The 1H and 13C NMR spectra were recorded on a Varian Unity Plus-500 instrument, and the chemical shifts are expressed in parts per million (δ) relative to tetramethylsilane. Infrared spectra were recorded with a Nicolet Magna IR TM 750 and Perkin Elmer 283B instruments. MS data were recorded with a JEOL JMS-AX 505 HA mass spectrometer. Electron impact mass spectra were obtained at 70 eV ionization energy. Vacuum chromatography was performed on Merck Kieselgel 60 (0.040-0.863 mm). TLC analyses were performed on TLC plates with Si gel 60 F254 (Merck) or ALUGRAM® SIL G/UV254 silica gel plates. The compounds were detected by their absorbance under UV254 and UV366, or with a charring solution (12 g of ceric ammonium sulfate dihydrate, 22.2 mL of concentrated H2SO4 and 350 g of ice). Solvents were distilled prior to use.
Plant material. The roots of P. radulifolium (HBK) H. Rob. & Brettell were collected in San Luis Potosí in 1995. Plant material was identified by Dr. Robert Bye and M. Sc. Edelmira Linares from the Instituto de Biología de la UNAM. The voucher Bye & Linares 20028 was deposited in the Ethnobotanical Collection, and the voucher Bye & Linares 20149 was deposited in the National Herbarium, of the Instituto de Biología de la UNAM.
Extraction and isolation. Previously we reported preparation of the n-hexane, CH2Cl2-EtOH (3:2) and MeOH extracts from the roots of this species, and the chemical analysis of the less polar residue [14]. The CH2Cl2-EtOH (3:2) extract (10 g) was loaded onto a column chromatography which was developed under reduced pressure using a gradient of n-hexane-EtOAc as elution system, to afford seven main fractions (named H to N). Fractions H and I were combined (31.2 mg), and this mixture was applied to a preparative TLC which was eluted with n-hexane-EtOAc (20:1), to yield maturinone [6b] (16, 5.6 mg), acetylmaturine [22] (17, 1.7 mg), and triacontanol (9 mg). Column rechromatography over silica gel of fraction J (350 mg) using n-hexane-EtOAc gradient as elution system afforded dimaturine [23] (18, 2.6 mg). Fractions K (15 mg) and L (1.3 g) were combined and subjected to Si-gel column chromatography using n-hexane-EtOAc (4:1) as elution system, affording decompostine [6d] (10, 896.5 mg). Fraction M (3.135 g) was chromatographed over Si gel using n-hexane-EtOAc gradient, to give several fractions. Some of these fractions were purified by column chromatography on Si gel using n-hexane-EtOAc gradient, leading to a mixture hydroxycacalolide [13a] (19) and epi-hydroxycacalolide [13a] (20, 3.3 mg), cacalone (2) and 6-epi-cacalone (3, 20 mg), radulifolin C [14] (7, 2.7 mg), radulifolin D (12, 7.1 mg), radulifolin E (ketodecompostine, 13, 2.2 mg). Column chromatography over Si gel of fraction N (2.56 g) using n-hexane-EtOAc as gradient elution system yielded β-sitosteryl 3-O-β-D-glucopyranoside, 14, 8.9 mg, radulifolin F (3-β-hydroxycacalone-3β-O-D-glucopyranoside, 14, 13.2 mg) and epi-radulifolin F (3-β-hydroxy-6-epi-cacalone-3β-O-D-glucopyranoside, 15, 26.3 mg) and β-D-glucopyranose. From the polar fractions of the metanolic extract (fractions Ñ to S) were identified O-methyl-1,2-dehydrocacalol [6e] (8), cacalol (1), decompostin (10), β-sitosterol, stigmasterol, cacalone (2) and 6-epi-cacalone (3), saccharose and β-D-glucopyranose.
Radulifolin D (12). 7.1 mg, yellow solid mp 122-124 °C, Rf: 0.226 (7:3 hex-AcOEt), mp 122-124 °C, [α]D = + 30.0 (c 0.05, MeOH); UV λmax (log ε) 207 (4.50), 245 (4.20), 277 (4.00), 336 (4.00); IR (CHCl3, cm−1): 3583, 3268, 2928, 2854, 1762, 1664, 1585, 1463, 1419, 1354, 1288, 1156, 1113, 996, 996, 918; 1H and 13C NMR data, see Tables 1 and 2; EIMS: C15H14O4, 258 [M+] (26), 243 (100), 240 (7), 215 (10), 201 (3), 187 (3), 85 (3), 157 (3), 135 (6), 128 (6), 115 (8), 109 (11), 91 (3), 77 (6), 55 (3), 43 (6).
Radulifolin E (ketodecompostin, 13). 4.4 mg, yellow solid mp 222-225 °C (lit. [6d] 220-221 °C), Rf: 0.413 (3:2 hex-AcOEt); Mp. 222-225 °C; UV λmax (log ε): 319.5 (3.95); 280.5 (3.58); 257 (3.71); 240.5 (3.65); 232 (3.66); 224.5 (3.65); 205.5 (3.72).; IR (CHCl3, cm−1): 3037, 2971, 2940, 1747, 1672, 1606, 1531, 1463, 1415, 1372, 1315, 1176, 1050, 1030, 983, 928; 1H and 13C NMR data, see Tables 1 and 2; EIMS: C17H18O5, 302 [M+] (10), 274 (0.5), 260 (64), 242 (100), 227 (15), 214 (9), 199 (14), 191 (24), 163 (5), 161 (5), 137 (20), 123 810), 115 (9), 109 (8), 91 (7), 77 (7), 65 (4), 53 (8), 43 (32), 41 (5).
Radulifolin F (3β-hydroxycacalone-3-O-β-D-glucopyranoside, 14). 13.2 mg, yellow oil; Rf: 0.295 (85:15 hex-AcOEt); IR (CHCl3, cm−1): 3401, 2936, 1660, 1619, 1603, 1535, 1459, 1445, 1421, 1363, 1162, 1079, 1035, 923, 887; 1H and 13C NMR data, see Tables 1 and 2; FABMS+: C21H28O9, 447 [M+ + Na] (58), 407 (20), 263 (48), 245 (100), 227 (72), 191 (24), 154 (31), 136 (28), 91 (20), 77 (19), 44 (17).
Epi-radulifolin F (3β-hydroxy-6-epi-cacalone-3-O-β-D-glucopyranoside, 15). 26.3 mg yellow oil; Rf: 0.295 (85:15 hex-AcOEt); IR (KBr, cm−1): 3411, 2930, 1654, 1614, 1535, 1451, 1422, 1369, 1256, 1224, 1201, 1164, 1078, 1038, 936, 888, 814, 623, 595, 532; 1H and 13C NMR data, see Tables 1 and 2; FABMS+: C21H28O9, 447 [M+ + Na] (100), 425 (17), 399 (8), 371 (38), 263 (49), 245 (44), 227 (26), 191 (21), 177 (35), 154 (59), 136 (51), 91 (26), 77 (25), 55 (24), 41 (25), 23 (58).
Biological evaluations. The methanol extract used for biological assays was obtained by direct maceration of the dried roots of the plant at room temperature (1 L per each 100 g) by 48 h two times. Male Wistar normoglycaemic rats of 60-65 days old, generally weighing 200-250 g, were used. The animals were housed under standard laboratory conditions and maintained on standard pellet diet and water ad libitum. Rats were placed in single cages with wire-net floors and deprived of food for 18 h before experimentation but allowed free access to tap water throughout. All experiments were carried out using 5 animals per group. Male Wistar rats were made diabetic by an intraperitoneal injection of streptozotocin (60 mg/kg) in citrate buffer, pH 6.3 [24]. Extracts were suspended in 0.05 % of Tween 80 in saline solution. Glibenclamide (10 mg/kg) was used as a hypoglycemic model drug [25]. All extracts were prepared freshly immediately before the experimentation and administered by intragastrical route at 30, 100 and 300 mg/kg. Control rats received the vehicle (0.05 % Tween 80) in the same volume (0.5 mL/100 g) by the same route. Blood samples were collected from caudal vein by means of a little incision in the end of the tail at 0, 1.5, 3, 5, 7 and 9 h after drug administration. Blood glucose concentration was estimated by enzymatic glucose oxidase method using a commercial glucometer (One Touch Basic I, Jonhsons-Johnsons). The percentage variation of glycemia for each group was calculated with respect to initial (0 h) level according to:
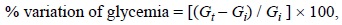
Where Giwas initial glycemia values and Gtwas the hypoglycemia value at +1.5, +3, +5, and +7 h, respectively [17b]. Statistical significance was estimated by analysis of variance (ANOVA) followed by Dunnett's test t. p < 0.05 implies significance.
Evaluation of antioxidant activity. The potential antioxidant activity of plant extracts and pure compounds was assessed on the basis of the scavenging activity of the stable 1,1-diphenyl-2-picrylhydrazyl (DPPH) free radical [26]. Reaction mixtures containing test samples (dissolved in ethanol, at 1, 10 and 100 µM) and DPPH ethanolic solution (66.66 µM) in ambar vials (4 mL) were stirred for 30 min, and absorbances were measured at 515 nm. Percent of inhibition by sample treatment was determined by comparison with a control group [27].
Acknowledgements
We thank Rocío Patiño, Beatriz Quiroz, María Isabel Chávez, Héctor Ríos, Luis Velasco, Javier Pérez-Flores, María Teresa Ramírez-Apan, and Antonio Nieto from the Instituto de Química de la UNAM for technical assistance; and Dr. Andrés Navarrete, Facultad de Química de la UNAM, for the use of certain research facilities and guidance in some hypoglycemic evaluations assays. Financial support by grants from the DGAPA-UNAM and PROMEP-UAEMor is gratefully acknowledged.
References
1. Linares, E.; Bye, R. A. J. Ethnopharmacol. 1987, 19, 153-186. [ Links ]
2. Bye, R. A. Econ. Bot. 1986, 40, 103-124. [ Links ]
3. Pérez, R. M.; Ocegueda, G. A.; Muñoz, J. L.; Ávila, J. G.; Morrow, W., W. J. Ethnopharmacol. 1984, 12, 253-262. [ Links ]
4. Sullivan, G. Vet. Hum. Toxicol. 1981, 23, 6-7. [ Links ]
5. Barkley, T. M.; Clark, B. L.; Funston, M. Compositae: Systematics. Proceedings of the International Compositae Conference, Kew, 1994. Hind, D. N. J., Ed. Royal Botanic Gardens, Kew, 1996; Vol. 1., pp. 613-620. [ Links ]
6. (a) Romo, J.; Joseph-Nathan, P. Tetrahedron 1964, 20, 2331-2337. [ Links ] (b) Correa, J.; Romo, J. Tetrahedron 1966, 22, 685-691. [ Links ] (c) Joseph-Nathan, P.; Morales, J. J.; Romo, J. Tetrahedron 1966, 22, 301-307. [ Links ] (d) Rodríguez-Hahn, L.; Guzmán, A.; Romo, J. Tetrahedron 1968, 24, 477-483. [ Links ] (e) Romo, J.; Rodríguez-Hahn, L.; Manjarrez, A.; Rivera, E.; Bellido, J. Bol. Inst. Quím. Univ. Nac. Autón. Méx. 1968, 20, 19-29. [ Links ] (f) Brown, M.; Thompson, R. H. J. Chem. Soc. 1969, 1184-1186. [ Links ] (g) Ruiz, R. M.; Correa, J.; Maldonado, L. A. Bull. Soc. Chim. Fr. 1969, 3612-3614. [ Links ] (h) Romo, J. Bol. Inst. Quím. Univ. Nac. Autón. Méx. 1969, 21, 92-96. [ Links ] (i) Kakisawa, H.; Inouye, Y.; Romo, J. Tetrahedron Lett. 1969, 1929-1932. [ Links ] (j) Samek, Z.; Harmatha, J.; Novotný, L.; Sorm, F. Coll. Czech. Chem. Comm. 1969, 34, 2792-2808. [ Links ] (k) Joseph-Nathan, P.; Negrete, M. C.; González, M. P. Phytochemistry 1970, 9, 1623-1628. [ Links ]
7. (a) Inouye, Y.; Uchida, Y.; Kakisawa, H. Chem. Lett. 1975, 1317-1318. [ Links ] (b) Yuste, F.; Walls, F. Aust. J. Chem. 1976, 29, 2333-2336. [ Links ] (c) Cásares, A.; Maldonado, L. A. Tetrahedron Lett. 1976, 2485-2488. [ Links ] (d) Yuste, F.; Díaz, E.; Walls, F.; Jankowski, K. J. Org. Chem. 1976, 41, 4103-4106. [ Links ] (e) Inouye, Y.; Uchida, Y.; Kakisawa, H. Bull. Chem. Soc. Jpn. 1977, 50, 961-966. [ Links ] (f) Huffman, J. W.; Pandian, R. J. Org. Chem. 1979, 44, 1851-1855. [ Links ] (g) Garofalo, A. W.; Litvak, J.; Wang, L.; Dubenko, L. G.; Cooper, R.; Bierer, D. E. J. Org. Chem. 1999, 64, 3369-3372. [ Links ]
8. Jiménez, M.; Cruz, R.; Valdés, J.; León, J.; Alarcón, G.; Sveshtarova, B. Rev. Latinoam. Quím. 1992, 23, 14-17. [ Links ]
9. Krasovaskaya, N. P.; Kulesh, N. I.; Denisenko, V.; Khim. Prirod. Soed. 1989, 643-645. [ Links ] Chem. Abst. 1990, 112, 95533u. [ Links ]
10. Anaya, A. L.; Hernández-Bautista, B. E.; Torres-Barragán, A.; León-Cantero, J.; Jiménez-Estrada, M. J. Chem. Ecol. 1996, 22, 393-406. [ Links ]
11. Román Ramos, R.; Lara, L. A.; Alarcón Aguilar, F. J.; Flores Sáenz, J. L. Arch. Med. Res. 1992, 23, 105-109. [ Links ]
12. Alarcón, F. J.; Román-Ramos, R.; Jiménez-Estrada, M.; Reyes-Chilpa, R.; González-Paredes, B.; Flores Sáenz, J. L. J. Ethnopharmacol. 1997, 55, 171-177. [ Links ]
13. (a) Inman, W. D.; Luo, J.; Jolad, S. D.; King, S. R.; Cooper, R. J. Nat. Prod. 1999, 62, 1088-1092. [ Links ] (b) Alarcón Aguilar, F. J.; Jiménez-Estrada, M.; Reyes Chilpa, R.; González Paredes, B.; Contreras Weber, C. C.; Román Ramos, R. J. Ethnopharmacol. 2000, 69, 207-215. [ Links ]
14. Garduño-Ramírez, M. L.; Trejo, A.; Navarro, V.; Bye, E.; Linares, E.; Delgado, G. J. Nat. Prod. 2001, 64, 432-435. [ Links ]
15. Naya, K.; Miyoshi, Y.; Mori, H.; Takai, K.; Nakanishi, M. Chem. Lett. 1976, 73-76. [ Links ]
16. This effect is evident using C6D5N as solvent, as described for the epimers 2 and 3 in reference 7d.
17. (a) Navarrete, A. Evaluación Farmacológica de Plantas Medicinales. En: Plantas Medicinales de México. Introducción a su Estudio. Editor: E. Estrada. Universidad Autónoma de Chapingo. México. 1996. pp. 255-268. [ Links ] (b) Sharma, S. R.; Dwivedi, S. K.; Swarup, D. J. Ethnopharmacol. 1997, 58, 39-44. [ Links ] (c) Fernando, M. R.; Wickramasinghe, S. M. D. N.; Thabrew, M. I.; Karunanyaka, E. H. J. Ethnopharmacol. 1989, 27, 7-14. [ Links ] (c) Trajanoski, Z.; Brunner, G. A.; Gferer, R. J.; Wach, P.; Pieber, T. R. Diabetes Care 1996, 19, 1412-1415. [ Links ]
18. Becker, M.; Newman, S.; Ismail-Beigi, F. Mol. Cell. Endocrinology 1996, 121, 165-170. [ Links ]
19. (a) Hennani, T.; Parihar, M. S. Ind. J. Phys. Pharm. 1998, 42, 440.452. [ Links ] (b) Anderson, S. M. Drug Develop. Res. 1999, 46, 67-79. [ Links ] (c) Sies, H. Oxidative Stress. Introductory Remarks. In: Sies, H., Ed. Academic Press. Orlando, Florida. 1985. pp. 1-8. [ Links ]
20. Lotina-Hennsen, B.; Roque Reséndiz, J. L.; Jiménez, M.; Aguilar, M. Z. Naturforsch. 1991, 46c, 777-780. [ Links ]
21. Meckes, M.; Garduño-Ramírez, M. L.; Marquina, S.; Álvarez, L. Rev. Soc. Quím. Méx. 2001, 45, 195-199. [ Links ]
22. Bohlmann, F.; Zdero, Ch.; Grenz, M. Chem. Ber. 1977, 110, 474-486. [ Links ]
23. (a) Joseph-Nathan, P.; Negrete M. C.; González, M. P. Phytochemistry 1970, 9, 1623-1628. [ Links ] (b) Bohlmann, F.; Zdero, Ch. Chem. Ber. 1970, 111, 3140-3145. [ Links ]
24. Bwititi, P.; Musabayane, C. T.; Nhachi, C. F. B. J. Ethnopharmacol. 2000, 69, 247-252. [ Links ]
25. Nicki, I.; Nicks, J. L.; Ashcroft, S. J. H. Biochem. J. 1990, 268, 713-718. [ Links ]
26. Cotelle, V.; Vernier, J. L.; Catteau, J. P.; Pommery, J.; Wallet, J. C.; Gaydou, E. M. Free Radical Biol. Med. 1996, 20, 35-43. [ Links ]
27. (a) Waffo, T. P.; Fauconneau, B.; Deffieux, G.; Huguet, F.; Vercauteren, J.; Merillon, J. M. J. Nat. Prod. 1998, 61, 655-657. [ Links ] (b) Rekka, E. A.; Kourounakis, A. P. Res. Comm. Mol. Pathol. Pharmacol. 1996, 92, 361-366. [ Links ]

