










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de la Sociedad Química de México
Print version ISSN 0583-7693
Rev. Soc. Quím. Méx vol.47 n.2 Ciudad de México Apr./Jun. 2003
Investigación
Synthesis, Structural, and Theoretical Study of New β-Heterosubstituted Captodative Olefins 1-Acetylvinyl Arenecarboxylates
Jorge A. Mendoza,1 Hugo A. Jiménez-Vázquez,1 Rafael Herrera,2 Jide Liu,1 and Joaquín Tamariz1*
1 Departamento de Química Orgánica, Escuela Nacional de Ciencias Biológicas, Instituto Politécnico Nacional, Prol. de Carpio y Plan de Ayala, 11340 México, D.F. México. Tel: (+5255) 5729-6300 / 62411; Fax: (+5255) 5396-3503. E-mail: jtamariz@woodward.encb.ipn.mx
2 Instituto de Investigaciones Quimicobiológicas, Universidad Michoacana de San Nicolás de Hidalgo, Edif. B-1, Ciudad Universitaria, Francisco J. Mujica S/N, 58066 Morelia, Mich., México.
Dedicated to Dr. Alfonso Romo de Vivar on the occasion of his 50th anniversary of relevant contributions in natural products chemistry at the Instituto de Química, UNAM.
Recibido el 10 de diciembre del 2002.
Aceptado el 6 de febrero del 2003.
Abstract
A new series of β-heterosubstituted captodative olefins 1-acetylvinyl arenecarboxylates (7a and 10a-10h) has been prepared, by introducing nitrogen or sulphur as heteroatoms substituted by alkyl and aryl groups. Three preparation methods were evaluated by modifying the leaving group of the starting material, as well as the nucleophilic character of the adding thiols. All of them were efficient and stereoselective, providing the desired alkenes in good yields and with the Z configuration of the double bond. Ab initio calculations (HF/6-31G*) of the FMOs, of some of the beta amino and bromo olefins, explained their experimental reactivity in Diels-Alder additions with respect to the unsubstituted olefin 1a. It also appears that the HOMO and LUMO energies of the beta sulphur analogues are governed by the particular electronic features of the sulphur atom, and that their very low reactivity before a diene is due to steric hindrance. A comparison between bond distances obtained by X-ray crystallography of different β-substituted and unsubstituted olefins seems to correlate with the delocalization effect of the heteroatom lone electron pair for the bromo and amino β-substituted olefins.
Keywords: Captodative olefin, structure, reactivity.
Resumen
Se describe la preparación de una nueva serie de olefinas captodativas β-heterosustituidas 1-acetilvinil arencarboxilatos (7a and 10a-10h), donde el heteroátomo es nitrógeno o azufre, sustituido por grupos alquilo y arilo. Se evaluaron tres métodos para su preparación, modificando el grupo saliente en el sustrato, y el carácter nucleofílico de los tioles que se adicionaron. Los tres métodos fueron eficientes y estereoselectivos, proporcionando los alquenos deseados en buenos rendimientos y con la configuración Z del doble enlace. El cálculo de orbitales moleculares frontera (ab initio, HF / 6-31G*) de algunas de las olefinas preparadas permitió explicar sus diferencias en reactividad experimental con respecto a la olefina no sustituida 1a. Así, se sugiere que las energías de los orbitales HOMO y LUMO de los análogos azufrados están gobernadas por las propiedades electrónicas particulares del átomo de azufre, y que su baja reactividad ante un dieno depende también del efecto estérico. Una comparación de los datos de cristalografía de rayos X entre distancias de enlace de diferentes olefinas β-sustituidas y la no sustituida parece correlacionarse con el efecto de deslocalización del par de electrones no compartidos del heteroátomo para las olefinas β-bromo y β-amino sustituidas.
Palabras clave: Olefina captodativa, estructura, reactividad.
Introduction
Captodative olefins have attracted particular attention in recent years, due to the opposite electronic demand and to the synthetic potential displayed by their geminally substituted functional groups [1]. We have shown that 1-acetylvinyl p-arenecarboxylates 1a-1c were highly reactive and selective in Diels-Alder [2] and 1,3-dipolar cycloadditions [3], and they also proved to be very useful synthons in natural product synthesis [4]. More recently, the alkyl 2-aroyloxy acrylates 2a-2b were prepared, showing also high reactivity and selectivity in Diels-Alder reactions [5]. With the aim of evaluating the effect of a third substituent in the double bond on the reactivity in [4+2] additions, compound 3a was prepared through a stereoselective synthetic route starting from 1a [6]. A series of amines 3b and thiols 3c and 3d were synthesized by treatment of 3a with the corresponding amines and thiols. In particular, enaminones are important organic intermediates [7], and have potential biological activity [8]. Moreover, other β-substituted captodative olefins have been prepared, showing interesting pericyclic behavior and synthetic usefulness [9].
Attempts to carry out the Diels-Alder cycloaddition of alkenes 3a-3d with dienes such as cyclopentadiene (4) and isoprene (5) were, however, unsuccessful, except for derivative 3a. The latter yielded adducts with diene 4 in an unexpectedly high exo stereoselectivity, and in comparable para/meta regioselectivity to that observed for olefin 1a with diene 5 [6].

The high reactivity and selectivity of captodative olefins in cycloaddition reactions is rather unexpected, since the electron-releasing effect of the aroyloxy group should decrease their reactivity in comparison with a dienophile or dipolarophile bearing only an electron-withdrawing group, such as methyl vinyl ketone (6) [10]. Structural and theoretical studies of olefin 1a revealed that the delocalization of the oxygen lone pair of the electron-donor group toward the π-sytem was inhibited by conformational restrictions [11]. In addition, FMO calculations suggested a dominant effect of the acetyl electron-withdrawing group on the polarization of the olefin [11]. However, the high regioselectivity shown by olefins 1 in 1,3-dipolar additions toward nitrones and nitrile oxides was better rationalized by DFT/HSAB theory [3c], showing the relevance of the electron-donor group in controlling the interaction of the cycloaddends. Therefore, electronic and structural factors should be taken into account to explain the reactivity and regiochemistry observed in both Diels-Alder and 1,3-dipolar reactions.
It is then relevant to evaluate the perturbation of the beta substituent in olefins 3 on the electronic and structural properties of these molecules and, in particular, on the double bond. Accordingly, we hereby report the preparation of a large series of new captodative olefins β-substituted with a new amine, and alkyl and aryl thiols. MO calculations were also carried out to assess the effect of the third substituent on the FMO energies and coefficients.
Results and discussion
Synthesis of β-heterosubstituted captodative olefins
The β-amino substituted olefin 7a was prepared by treating α-bromoalkene 3a with amine 8a, as an extension of the method used for the preparation of amino derivatives 3b [6] (Fig. 1). Thus, addition of N,N-methylphenyl amine (8a), in methylene chloride at 10 °C for 30 min, satisfactorily led to the desired -substituted olefin 7a in 62 % yield. The Z configuration of the double bond was established through NOE experiments. Irradiation of proton H-4 resulted in enhancement of the signal for the acetyl group.

In the previous report, the synthesis of β-thio (β-sulfanyl) olefins was limited to 3c and 3d by treatment of 3a with the corresponding thiol in the presence of triethylamine in DMF at room temperature [6]. The use of some other alkyl mercaptanes and aryl thiophenols under these conditions was not as efficient. Therefore, three additional methodologies were investigated to improve the yields. The first method involved the addition of the sodium salt of the thiol (9a-9c) to 3a (Fig. 2), leading to products 10a-10c in high yields and under mild conditions (Table 1, entries 1-3). When thiols 9d-9g were used, more severe conditions were applied in order to improve the preparation of olefins 10b-10e (entries 4-7). The yields were high even for the bulky thiol 9f. Under the same reaction conditions, substituted thiophenols 9h-9j reacted with 3a to give alkenes 10f-10h also in good yields (entries 8-10).
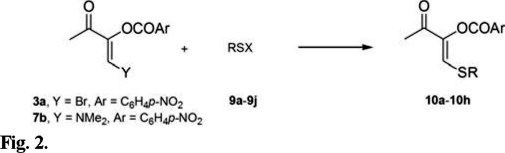
The leaving group Y at the starting alkenes was also evaluated. Instead of the bromine atom of olefin 3a, the dimethylamino group was used, due to its known aptitude as a leaving group [12]. In these cases the optimum reaction conditions were found to be similar to those employed for olefin 3a, that is, reacting olefin 7b in the presence of thiols 9d-9j and tri-ethylamine as base, and heating from 0 °C up to 120 °C for 3 h (Table 2). The aliphatic thiols 9d-9g provided compounds 10b-10e in quite lower yields than those obtained when olefin 3a was used (Table 2, entries 1-4 vs. Table 1, entries 4-7); however, the yields increased for the preparation of 10g-10h when 7b was treated with thiophenols 9i-9j (Table 2, entries 6-7 vs. Table 1, entries 9-10).

For both olefins, 3a and 7b, there were no significant differences in reactivity or yields to furnish the β-substituted alkenes 10 with the para substituted thiophenols 9h-9j (Table 1, entries 8-10 and Table 2, entries 5-7), considering that the temperature and the reaction time were comparable, regardless of the substituent.
It is interesting that sodium thiolates 9a-9c reacted with olefin 3a to give the 1,4-addition products 10a-10c in good yields, instead of providing the hydrolisis products by addition to the p-nitrobenzoyloxy group, as observed when the corresponding alcohols were used. As expected, this behavior illustrates again the known greater softness of the sulphur atom with respect to the oxygen atom.
The Z configuration of the double bond is maintained in all of the new olefins 10a-10h, as observed for the β-amino alkene 7a, indicating that it is largely more stable than the E configuration [6]. It is likely that the higher stability of the Z configuration is associated to destabilizing steric interactions found between the acetyl group and the beta substituent in the opposite E configuration. Such interactions may inhibit the effective conjugation of the enone moiety, and the delocalization of the heteroatom lone electron pair through the π system [6, 13].
The configuration of the double bond and the planar conformation of the enone moiety was confirmed by single crystal X-ray crystallography of olefin 10f (Fig. 3). It shows that both conjugated moieties, the enone and p-nitrobenzoyl groups, are in a quasi-orthogonal conformation. This agrees with those structures obtained for other analogues [5, 6, 11], as well as in the s-trans conformation of the enone. Interestingly, the p-bromophenyl ring lies out of the plane formed by the enone π-system.

FMO calculations of β-heterosubstituted captodative olefins
Frontier molecular orbital (FMO) theory has proven to be a reliable model to predict reactivity and regioselectivity in Diels-Alder [14] and 1,3-dipolar cycloadditions [15]. It has been able to explain the behavior of substituted olefins as dienophiles in Diels-Alder additions. For instance, the rate increases when the dienophile bears electron-withdrawing groups, whereas it decreases with dienophiles bearing electron-donating groups [16].
FMO theory has also been useful in explaining the reactivity and regioselectivity of captodative olefins with dienes such as isoprene (5) [2a, 11], showing that the interaction was controlled by normal electron demand (NED), i.e. the HOMO-diene/LUMO-dienophile interaction was the energetically most favorable. Furthermore, these results were supported by experimental measurement of ionization energies (IEs) and vertical attachment energies (VAEs) of 1a, whose relative values fit well with the calculated HOMO and LUMO energies, respectively [11].
Therefore, the electronic effect produced by the substituent in the beta position of the captodative olefins 3, 7, and 10 on their behavior in Diels-Alder reactions could be evaluated by calculating the FMO energies of these molecules, and correlating them with those of the corresponding FMOs of a diene such as 5.
Table 3 summarizes the calculated (HF/6-31G*) FMO energies of bromo and amino alkenes 3a, 7a, and 7b, as well as the thio alkenes 10a, 10f, and 10g. The geometries were optimized with the same basis set, showing that the most stable geometry for 3a, 7a, and 7b, corresponded to the s-trans conformation of the enone moiety, and the non-planar conformation of the p-nitrobenzoyloxy group. However, only for thioether 10g, the enone s-trans conformation was more stable, while olefins 10a and 10f were the exception, since the s-cis conformer for the enone conjugate system was slightly more stable (0.72 kcal/mol for 10a, and 0.24 kcal/mol for 10f), which is not in agreement with the X-ray structure of 10f. Even though these energy differences are negligible, and, practically, both conformations are isoenergetic, the FMO energy values listed in Table 3 corresponded to those obtained for the most stable confomation in every molecule. The coefficient differences follow a similar trend in the other conformers.

As expected for the amino substituted olefins 7a and 7b, when an electron-releasing group is introduced into the double bond of the captodative olefin, both HOMO and LUMO should increase in energy (Table 3, entry 1 vs. entries 3 and 4). However, for the rest of the olefins, only the HOMO was energetically destabilized, since the LUMO energy of olefins 3a, 10f, and 10g was lower than that of the unsubstituted olefin 1a. It is likely that the effect of the heteroatom on the energy of the FMOs be the result of an interplay of different factors. The delocalization of the lone electron pair of bromine and sulphur toward the π-conjugated enone system will be less efficient than that of the nitrogen atom, due to the differences in electronic configuration. The inductive effect of these electronegative heteroatoms may increase the electron-withdrawing effect of the beta substituent on the electron density of the double bond [17].
The delocalization of the lone electron pair in enaminone 7b was reflected in a shortening of the C(1)-N and C(2)-C(3) bonds, and by an increase of the C(3)=O(4) and C(1)=C(2) bond lengths [6], as observed for other analogues by X-ray diffraction [18]. Similarly, significant delocalization appears in the bromo olefin 3a, since the C(2)-C(3) bond length is shorter and the C(1)=C(2) bond is longer than the corresponding bond lengths of 1a and of the average values taken from X-ray data of similar functional groups [19] (Table 4). In contrast, the X-ray structure of 10f (Fig. 2) provides bond distances similar to those for the β-unsubstituted olefin and for the non-delocalized enone [19] (Table 4). It is noteworthy that the bond distances between the atoms of the aroyloxy group are not really perturbed by changes in the heteroatom at the beta position. This supports the idea of a non significant interaction between the lone pair of the oxygen atom of the electron donor group and the double bond, at least, in the crystalline state.
According to data in Table 3, one could anticipate a higher reactivity of olefins 3a, 10f, and 10g with respect to 1a in Diels-Alder cycloadditions with diene 5, since the HOMO-diene / LUMO-dienophile energy gaps for the former dienophiles are smaller than that found for the latter. This expectation is only partially in agreement with the observed reactivity, since olefin 3a added to 5 under thermal and Lewis acid catalytic conditions similar to those used for 1a in shorter reaction times [6]. However, thioalkene 3d, which is analogous to olefins 10, failed to react even under catalysis. As predicted, the beta amino substituted alkene 7b was less reactive than 1a, being unable to react with cyclopentadiene (12) [6], which is considered a very reactive diene.
The unreliable prediction of the reactivity of olefins 10 by FMO theory may be attributed to steric hindrance [20], which would counterbalance the electronic effect, as suggested previously by establishing a correlation between the reaction rate and LUMO energies of olefins 1a and 6 [11]. In addition, it has been found that the FMO model fails to account for the regioselectivity in Diels-Alder reactions with trisusbtituted dienophiles or with phenylthiosubstituted dienes [21], or in 1,3-dipolar cycloadditions of nitrile oxides and nitrones with olefins such as 1a [3c].
It is noteworthy that, under thermal and catalyzed conditions, the regioselectivity found in the Diels-Alder addition of dienophile 3a with isoprene (5) was similar to that observed with the β-unsubstituted olefin 1a [2a, 6]. From the FMO viewpoint, regioselectivity can be predicted on the basis of the atomic coefficient differences for the appropriate frontier orbital interaction: HOMO-diene/LUMO-dienophile, under NED control [14a, 14b]. It can be observed from Table 5 that, for olefin 3a, the relative magnitude of the LUMO coefficient in the monosubstituted terminus of the double bond is larger than that of the geminally disubstituted carbon. In comparison with the LUMO of olefin 1a, the difference in coefficients C1 and C2 for this olefin and bromo olefin 3a are analogous, hence a comparable regioselectivity should be observed, which is, indeed, experimentally found. Thus, for diene 5, which has the HOMO largest coefficient in carbon C-1, a preferred interaction with the largest coefficient on the double bond of the dienophile agrees with the para orientation as the major regioisomer.
Polarization of the π-system in the HOMO for the amino olefins 7a and 7b, and for the thio olefins 10 is towards the substituted captodative carbon of the double bond (Table 5). The larger difference in coefficients (ΔCi) of these alkenes with respect to olefin 1a reflects the electron-donor effect of the heteroatom in beta position. In contrast, the opposite polarization is found for the LUMO, where the larger coefficient is located in the monosubstituted carbon atom (C1) of the double bond.
Conclusions
The stereoselective synthesis of new β-heteroatom substituted captodative olefins, including amino compound 7a, and sulphur derivatives 10a-10h, was feasible through three analogous routes. The common feature among them involved the replacement of a leaving group at the beta position of the olefin by the corresponding amino or thio compound. Both bromo and dimethylamino were efficient as the leaving groups in the starting activated substrates 3a and 7b, respectively. The former underwent nucleophilic attack of alkyl and aryl thiols or the sodium salt of some of them in good yields. The Z configuration of the double bond was established by NMR and X-ray crystallography. A comparison between bond distances by X-ray crystallography of different β-substituted and unsubstituted olefins seems to correlate with the delocalization effect of the heteroatom lone electron pair for the bromo and amino β-substituted olefins.
Ab initio calculations of FMOs of trisubstituted amino olefins 7a and 7b showed an increase of HOMO and LUMO energies with respect to the unsubstituted 1a, as expected for the perturbation of the π-orbital by an electron-donating group. The presence of a bromine atom and alkyl and aryl thio groups in olefins 3a, and 10a, 10f, and 10g, however, produced an increase of the HOMO energy and a stabilization of the LUMO. Based on these results, the predicted reactivity of the Diels-Alder additions with diene 5 agrees with experiment only for alkenes 3a, 7a and 7b. The regioselectivity observed for the cycloaddition of olefin 3a is also explained by FMO theory. Therefore, these calculations clearly indicate a significant perturbation of the double bond of the captodative olefin by the third substituent in the beta position.
Experimental section
General. Melting points (uncorrected) were determined with an Electrothermal capillary melting point apparatus. IR spectra were recorded on a Perkin-Elmer 1600 spectrophotometer. 1H and 13C NMR spectra were obtained on a Varian Gemini-300 (300 MHz and 75.4 MHz), and Brucker DMX-500 (500 MHz and 125 MHz) instruments, with CDCl3 as solvent and TMS as internal standard. The mass spectra (MS) were taken on a Hewlett-Packard 5971A spectrometer. X-Ray analyses were collected using Mo Kα radiation (graphite crystal monochromator, λ = 0.71073 Å). Microanalyses were performed by M-H-W Laboratories (Phoenix, AZ). Analytical thin-layer chromatography was carried out using E. Merck silica gel 60 F254 coated 0.25 plates, visualizing by long- and short-wavelength UV lamp. All air moisture sensitive reactions were carried out under nitrogen using oven-dried glass-ware. DMF was freshly distilled and received on molecular sieves (4 Å), and methylene chloride from calcium hydride, prior to use. Triethylamine was freshly distilled from NaOH. All other reagents were used without further purification. Compounds 3a and 7b were prepared as described previously [6].
(Z)-4-(N,N-Methylphenylamino)-3-(4-nitrobenzoyloxy)-3-buten-2-one (7a). To a solution of 1.0 g (3.18 mmol) of 3a in CH2Cl2 (25 mL), at 10 °C, 0.443 g (4.14 mmol) of 8a were added, and the mixture was stirred for 30 min. The reaction mixture was diluted with CH2Cl2 (50 mL), and was washed with a cold 5 % aqueous solution of HCl (2 × 25 mL), and a cold saturated solution of NaCl (2 × 30 mL). The organic layer was dried (MgSO4), and the solvent was evaporated under vacuum. The residue was successively purified by flash column chromatography on silica gel treated with 10 % of triethylamine (20 g, hexane/EtOAc, 90:10), and by radial chromatography (hexane/CH2Cl2, 90:10). The solid was recrystallized (hexane/CH2Cl2, 20:80) to give 0.67 g (62 %) of 7a as pale brown crystals: Rf0.14 (hexane/EtOAc, 8:2); mp 141-143 °C; IR (CH2Cl2) 1741, 1621, 1591, 1529, 1495, 1350, 1316, 1244, 1096, 896 cm−1; 1H NMR (300 MHz, CDCl3) δ 2.24 (br s, 3H, CH3CO), 3.45 (s, 3H, CH3N), 6.99-7.04 (m, 1H, Ph-H), 7.13-7.16 (m, 2H, Ph-H), 7.22-7.27 (m, 2H, Ph-H), 7.48 (br s, 1H, HC=), 8.03-8.06 (m, 2H, Ar-H), 8.21-8.24 (m, 2H, Ar-H); 13C NMR (75.4 MHz, CDCl3) δ 24.3 (CH3CO), 42.1 (CH3N), 122.7, 123.2, 125.8, 126.8 (C-3), 129.2, 130.9 (C-4), 131.1, 134.3, 145.7, 150.6, 162.7 (ArCO2), 187.0 (CH3CO). Anal. Calcd for C18H16N2O5: C, 63.52; H, 4.74; N, 8.23. Found: C, 62.75; H, 4.58; N, 7.71.
General Procedures for the Preparation of Olefins 10a-10h. Method A. To a mixture of 1.0 g (3.18 mmol) of 3a and 1.31 mol equiv. of the sodium salt of the corresponding thiol 9a-9c, at 0 °C, anhydrous DMF (10 mL) was added, and the mixture was stirred for 30 min. The solution was concentrated under vacuum, the reaction crude was diluted with CH2Cl2 (50 mL), and was washed with a cold 5 % aqueous solution of HCl (2 × 25 mL), and a cold saturated solution of NaCl (2 × 30 mL). The organic layer was dried (MgSO4), and the solvent was evaporated under vacuum. The residue was successively purified by column chromatography on silica gel treated with 10 % of triethylamine (30 g/1 g of crude, hexane/ EtOAc, 90:10), and by radial chromatography (hexane/CH2Cl2, 80:20).
Method B. To a solution of 1.0 g (3.18 mmol) of 3a in anhydrous DMF (20 mL) 1.31 mol equiv. of the corresponding thiol 9d-9j were added at 20 °C. The mixture was stirred and cooled down to 0 °C, and a solution of 0.42 g (4.16 mmol) of triethylamine in anhydrous DMF (3 mL) was added dropwise. The mixture was maintained at the same temperature for 1 h, then warmed up to 120 °C for 1 h, and cooled down to 20 °C for 1 h. The solution was concentrated under vacuum, the reaction crude was diluted with CH2Cl2 (50 mL), and washed with a cold 5 % aqueous solution of HCl (2 × 25 mL), and a cold saturated solution of NaCl (2 × 30 mL). The organic layer was dried (MgSO4), and the solvent was evaporated under vacuum. The residue was successively purified by column chromatography on silica gel treated with 10 % of triethylamine (30 g/1 g of crude, hexane/EtOAc, 90:10), and by radial chromatography (hexane/CH2Cl2, 90:10). For the solid products, the recrystallization was carried out from hexane/CH2Cl2, 10:90.
Method C. To a solution of 1.0 g (3.59 mmol) of 7b in anhydrous DMF (25 mL) 1.3 mol equiv. of the corresponding thiol 9d-9j were added at 20 °C. The mixture was stirred and cooled down to 0 °C, and a solution of 0.472 g (4.67 mmol) of triethylamine in anhydrous DMF (3 mL) was added dropwise. The mixture was maintained at the same temperature for 1 h, then warmed up to 120 °C for 1 h, and cooled down to 20 °C for 1 h. The solution was concentrated under vacuum, the reaction crude was diluted with CH2Cl2 (50 mL), and was washed with a cold 5 % aqueous solution of HCl (2 × 25 mL), and a cold saturated solution of NaCl (2 × 30 mL). The organic layer was dried (MgSO4), and the solvent was evaporated under vacuum. The residue was successively purified by column chromatography on silica gel treated with 10 % of triethylamine (30 g/1 g of crude, hexane/EtOAc, 80:20), and by radial chromatography (hexane/CH2Cl2, 80:20). For the solid products, the recrystallization was carried out from hexane/CH2Cl2, 10:90.
(Z)-4-Methylsulfanyl-3-(p-nitrobenzoyloxy)-3-buten-2-one (10a). According to method A with 0.291 g (4.16 mmol) of 9a, afforded 0.76 g (85 %) of 10a as a pale yellow oil: Rf0.21 (hexane/EtOAc, 8:2); IR (CH2Cl2) 1746, 1675, 1589, 1530, 1347, 1248, 1093, 895 cm−1; 1H NMR (300 MHz, CDCl3) δ 2.34 (s, 3H, CH3CO), 2.50 (s, 3H, CH3S), 7.39 (s, 1H, HC=), 8.32-8.34 (m, 4H, Ar-H); 13C NMR (75.4 MHz, CDCl3) δ 17.3 (CH3S), 24.7 (CH3CO), 123.7, 131.4, 134.1, 136.9 (C-4), 142.3 (C-3), 151.1, 161.7 (ArCO2), 187.1 (CH3CO); MS (70 eV) 281 (M+, 4), 150 (100), 134 (22), 120 (15), 104 (22), 92 (11), 76 (14).
(Z)-4-Ethylsulfanyl-3-(p-nitrobenzoyloxy)-3-buten-2-one (10b). According to method A with 0.349 g (4.16 mmol) of 9b, afforded 0.84 g (90 %) of 10b as a pale yellow oil. According to method B with 0.258 g (4.16 mmol) of 9d, gave 0.73 g (78 %) of 10b. According to method C with 0.289 g (4.67 mmol) of 9d, furnished 0.83 g (74%) of 10b: Rf0.22 (hexane/EtOAc, 8:2); IR (CH2Cl2) 1746, 1675, 1588, 1529, 1423, 1348, 1275, 1246, 1217, 1049, 1013 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.43 (t, J = 7.1 Hz, 3H, CH3CH2S), 2.35 (s, 3H, CH3CO), 2.92 (q, J = 7.1 Hz, 3H, CH3CH2S), 7.49 (s, 1H, HC=), 8.45-8.47 (m, 4H, Ar-H); 13C NMR (75.4 MHz, CDCl3) δ 15.4 (CH3CH2S), 24.7 (CH3CO), 28.4 (CH3CH2S), 123.6, 131.4, 134.1, 135.1 (C-4), 142.5 (C-3), 151.0, 161.7 (ArCO2), 187.1 (CH3CO). Anal. Calcd for C13H13NO5S: C, 52.87; H, 4.44. Found: C, 52.80; H, 4.68.
(Z)-4-Isopropylsulfanyl-3-(p-nitrobenzoyloxy)-3-buten-2-one (10c). According to method A with 0.408 g (4.16 mmol) of 9c, afforded 0.88 g (90 %) of 10c as a pale yellow oil. According to method B with 0.358 g (4.16 mmol) of 9e, gave 0.80 g (81 %) of 10c. According to method C with 0.402 g (4.67 mmol) of 9e, furnished 0.90 g (77 %) of 10c: Rf0.30 (hexane/EtOAc, 8:2); IR (CH2Cl2) 1746, 1674, 1587, 1530, 1453, 1427, 1348, 1314, 1218, 1157, 1092, 1015, 958, 898, 870, 847 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.38 (d, J = 7.3 Hz, 6H, (CH3)2CHS), 2.35 (s, 3H, CH3CO), 3.33 (sept, J = 7.3 Hz, 3H, (CH3)2CHS), 7.50 (s, 1H, HC=), 8.38-8.36 (m, 4H, Ar-H); 13C NMR (75.4 MHz, CDCl3) δ 23.4 ((CH3)2CHS), 24.5 (CH3CO), 38.4 ((CH3)2CHS), 123.4, 131.1, 133.9, 134.1 (C-4), 141.9 (C-3), 150.7, 161.5 (ArCO2), 187.0 (CH3CO). Anal. Calcd for C14H15NO5S: C, 54.36; H, 4.89. Found: C, 54.30; H, 5.00.
(Z)-4-tert-Butylsulfanyl-3-(p-nitrobenzoyloxy)-3-buten-2-one (10d). According to method B with 0.375 g (4.13 mmol) of 9f, afforded 0.84 g (82 %) of 10d as a pale yellow oil. According to method C with 0.514 g (4.67 mmol) of 9f, furnished 0.86 g (70%) of 10d: Rf0.26 (hexane/EtOAc, 8:2); IR (CH2Cl2) 1744, 1673, 1584, 1529, 1349, 1248, 1093, 769, 751 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.48 (s, 9H, (CH3)3CS), 2.35 (s, 3H, CH3CO), 7.60 (s, 1H, HC=), 8.33 (br s, 4H, Ar-H); 13C NMR (75.4 MHz, CDCl3) δ 24.8 (CH3CO), 31.1 ((CH3)3CS), 45.7 ((CH3)3CS), 123.7, 131.4, 131.6 (C-4), 134.2, 142.3 (C-3), 151.0, 162.4 (ArCO2), 198.8 (CH3CO). Anal. Calcd for C15H17NO5S: C, 55.71; H, 5.30. Found: C, 55.50; H, 5.30.
(Z)-4-Benzylsulfanyl-3-(p-nitrobenzoyloxy)-3-buten-2-one (10e). According to method B with 0.516 g (4.13 mmol) of 9g, afforded 1.02 g (90 %) of 10e as a pale yellow oil. According to method C with 0.579 g (4.67 mmol) of 9g, furnished 1.18 g (87 %) of 10e: Rf0.28 (hexane / EtOAc, 8:2); IR (CH2Cl2) 1740, 1677, 1606, 1530, 1348, 1248, 1093 cm−1; 1H NMR (300 MHz, CDCl3) δ 2.25 (s, 3H, CH3CO), 4.08 (s, 2H, CH2Ph), 7.27-7.46 (m, 6H, HC=, Ph-H), 8.28-8.39 (m, 4H, Ar-H); 13C NMR (75.4 MHz, CDCl3) δ 24.7 (CH3CO), 38.0 (CH2Ph), 123.7, 128.2, 129.0, 129.1, 131.4, 134.0, 134.2 (C-4), 136.7, 142.4 (C-3), 150.9, 161.7 (ArCO2), 187.1 (CH3CO).
(Z)-4-(4-Bromophenylsulfanyl)-3-(4-nitrobenzoyloxy)-3-buten-2-one (10f). According to method B with 0.786 g (4.16 mmol) of 9h, afforded 1.29 g (96 %) of 10f as pale yellow crystals. According to method C with 0.883 g (4.67 mmol) of 9h, furnished 1.45 g (91 %) of 10f: mp 174-175 °C; Rf0.16 (hexane/EtOAc, 8:2); IR (CH2Cl2) 1747, 1679, 1531, 1419, 1348, 1243, 1090, 1008 cm−1; 1H NMR (300 MHz, CDCl3) δ 2.35 (s, 3H, CH3CO), 7.36-7.40 (m, 2H, Ar-H), 7.46 (s, 1H, HC=), 7.51-7.55 (m, 2H, Ar-H), 8.30-8.38 (m, 4H, Ar-H); 13C NMR (75.4 MHz, CDCl3) δ 24.9 (CH3CO), 123.6, 123.7, 130.8, 131.5, 132.9, 133.1, 133.7, 134.1 (C-4), 142.6 (C-3), 151.2, 161.7 (ArCO2), 187.3 (CH3CO). Anal. Calcd for C17H12BrNO5S: C, 48.36; H, 2.86; N 3.32; S, 7.58. Found: C, 48.04; H, 2.80; N, 3.29; S, 7.41.
(Z)-4-(4-Methoxyphenylsulfanyl)-3-(4-nitrobenzoyloxy)-3-buten-2-one (10g). According to method B with 0.582 g (4.16 mmol) of 9i, afforded 0.95 g (80 %) of 10g as pale yellow crystals. According to method C with 0.654 g (4.67 mmol) of 9i, furnished 1.25 g (93 %) of 10g: mp 122-123 °C; Rf0.20 (hexane/EtOAc, 8:2); IR (CH2Cl2) 1743, 1674, 1592, 1546, 1531, 1492, 1421, 1346, 1264, 1093 cm−1; 1H NMR (300 MHz, CDCl3) δ 2.33 (s, 3H, CH3CO), 3.82 (s, 3H, MeO), 6.88-6.94 (m, 2H, Ar-H), 7.44-7.49 (m, 2H, Ar-H), 7.45 (s, 1H, HC=), 8.34 (s, 4H, Ar-H); 13C NMR (75.4 MHz, CDCl3) δ 24.8 (CH3CO), 55.4 (CH3O), 115.3, 122.0, 123.6, 131.5, 134.0, 134.2, 137.2 (C-4), 141.5 (C-3), 150.9, 160.7, 161.7 (ArCO2), 187.2 (CH3CO). Anal. Calcd for C18H15NO6S: C, 57.90; H, 4.05; N, 3.75; S, 8.59. Found: C, 58.08; H, 4.12; N 3.74; S, 8.70.
(Z)-4-(4-Methylphenylsulfanyl)-3-(4-nitrobenzoyloxy)-3-buten-2-one (10h). According to method B with 0.516 g (4.16 mmol) of 9j, afforded 0.92 g (81 %) of 10h as pale yellow crystals. According to method C with 0.58 g (4.67 mmol) of 9j, furnished 1.15 g (90 %) of 10h: mp 113-115 °C; Rf0.22 (hexane/EtOAc, 8:2); IR (CH2Cl2) 1747, 1677, 1589, 1531, 1348, 1091, 897, 847, 807 cm−1; 1H NMR (500 MHz, CDCl3) δ 2.33 (s, 3H, CH3CO), 2.37 (s, 3H, MeAr), 7.20-7.22 (m, 2H, Ar-H), 7.39-7.41 (m, 2H, Ar-H), 7.50 (s, 1H, HC=), 8.34 (s, 4H, Ar-H); 13C NMR (125 MHz, CDCl3) δ 21.1 (CH3Ar), 24.8 (CH3CO), 123.7, 128.2, 130.5, 131.5, 131.9, 134.0, 136.3 (C-4), 138.5, 141.9 (C-3), 151.0, 161.7 (ArCO2), 187.3 (CH3CO). Anal. Calcd for C18H15NO5S: C, 60.49; H, 4.23. Found: C, 60.27; H, 4.50.
Single-Crystal X-Ray Crystallography [22]. Olefin 10f was obtained as pale yellow crystals. These were mounted in glass fibers. Crystallographic measurements were performed on a Siemens P4 diffractometer with Mo Kα radiation (λ = 0.7107 Å; graphite monochromator) at room temperature. Two standard reflections were monitored periodically; they showed no change during data collection. Unit cell parameters were obtained from least-squares refinement of 26 reflections in the range 2 < 2Θ < 20°. Intensities were corrected for Lorentz and polarization effects. No absorption correction was applied. Anisotropic temperature factors were introduced for all non-hydrogen atoms. Hydrogen atoms were placed in idealized positions and their atomic coordinates refined. Unit weights were used in the refinement. The structure was solved using SHELXTL on a personal computer [23]. Data of 10f: Formula: C17H12BrNO5S; molecular weight: 422.25; cryst. syst.: monoclinic; space group: P21/c; unit cell parameters: a, 6.4121 (9), b, 12.8041 (11), c, 20.998 (2) (Å); α, 90, β, 94.884 (9), γ, 90 (deg); temp. (°K): 293 (2); Z: 4; No. of reflections collected: 4641; no. of independent reflections: 3335; no. of observed reflections: 3290; R: 0.0429; GOF: 1.012.
Calculations. The ab initio SCF/HF calculations were carried out with the 6-31G* basis sets using Gaussian 94 [24] and MacSpartan [24]. Geometries were fully optimized by the AM1 semiempirical method [25] and these were employed as starting point for optimization, at the 6-31G* level.
Acknowledgments
We thank Fernando Labarrios for his help in spectrometric measurements. J.T. would like to acknowledge DEPI/IPN (Grants 921769 and 200410) and CONACyT (Grants 1570P and 32273-E) for financial support. H.A.J.-V. thanks CONACyT (Grant 3251P) for financial support. J.M. and R.H. are grateful to CONACyT (Grants 86038 and 91187) for graduate fellowships, to PIFI-IPN program for a scholarship, and to the Ludwig K. Hellweg Foundation for a partial scholarship. J.L. is grateful to Secretaría de Relaciones Exteriores, México (Grant DAC-III 811.5/(510)/137) for a research fellowship. J.T. and H.A.J.-V. are fellows of the EDD/IPN and COFAA/IPN programs.
References and notes
1. (a) Viehe, H. G.; Janousek, Z.; Merényi, R.; Stella, L. Acc. Chem. Res. 1985, 18, 148-154. [ Links ] (b) Cativiela, C.; Fraile, J. M.; García, J. I.; Mayoral, J. A.; Pires, E.; Royo, A. J.; Figueras, F.; de Ménorval, L. C. Tetrahedron 1993, 49, 4073-4084. [ Links ] (c) Boucher, J.-L.; Stella, L. Tetrahedron 1986, 42, 3871-3885. [ Links ] (d) Seerden, J.-P. G.; Scheeren, H. W. Tetrahedron Lett. 1993, 34, 2669-2672. [ Links ] (e) Döpp, D.; Libera, H. Tetrahedron Lett. 1983, 24, 885-888. [ Links ] (f) Rulev, A. Y. Russ. Chem. Rev. 2002, 71, 195-221. [ Links ] (g) Seneci, P.; Leger, I.; Souchet, M.; Nadler, G. Tetrahedron 1997, 53, 17097-17114. [ Links ] (h) Moody, C. J.; Hughes, R. A.; Thompson, S. P.; Alcaraz, L. Chem. Commun. 2002, 1760-1761. [ Links ] (i) Kozmin, S. A.; Iwama, T.; Huang, Y.; Rawal, V. H. J. Am. Chem. Soc. 2002, 104, 4628-4641. [ Links ] (j) Ferreira, P. M. T.; Maia, H. L. S.; Monteiro, L. S. Tetrahedron Lett. 2002, 43, 4491-4493. [ Links ] (k) Ferreira, P. M. T.; Maia, H. L. S.; Monteiro, L. S. Tetrahedron Lett. 2002, 43, 4495-4497. [ Links ] (l) Huang, C.-G.; Chang, B.-R.; Chang, N.-C. Tetrahedron Lett. 2002, 43, 2721-2723. [ Links ]
2. (a) Reyes, A.; Aguilar, R.; Muñoz, A. H.; Zwick, J.-C.; Rubio, M.; Escobar, J.-L.; Soriano, M.; Toscano, R.; Tamariz, J. J. Org. Chem. 1990, 55, 1024-1034. [ Links ] (b) Aguilar. R.; Reyes. A.; Tamariz, J.; Birbaum, J.-L. Tetrahedron Lett. 1987. 28. 865-868. [ Links ] (c) García de Alba, O.; Chanona, J.; Delgado, F.; Zepeda, G.; Labarrios, F.; Bates, R. W.; Bott, S.; Juaristi, E.; Tamariz, J. Anal. Quím., Int. Ed. 1996, 92, 108-117. [ Links ]
3. (a) Nagarajan, A.; Zepeda, G.; Tamariz, J. Tetrahedron Lett. 1996, 37, 6835-6838. [ Links ] (b) Jiménez, R.; Pérez, L.; Tamariz, J.; Salgado, H. Heterocycles 1993, 35, 591-598. [ Links ] (c) Herrera, R.; Nagarajan, A.; Morales, M. A.; Méndez, F.; Jiménez-Vázquez, H. A.; Zepeda, L. G.; Tamariz, J. J. Org. Chem. 2001, 66, 1252-1263. [ Links ]
4. (a) Andrade, R. M.; Muñóz, A. H.; Tamariz, J. Synth. Commun. 1992, 22, 1603-1609. [ Links ] (b) Orduña, A.; Zepeda, G.; Tamariz, J. Synthesis 1993, 375-377. [ Links ] (c) Dienes, Z.; Vogel, P. J. Org. Chem. 1996, 61, 6958-6970. [ Links ] (d) Ochoa, M. E.; Arias, A. S.; Aguilar, R.; Delgado, F.; Tamariz, J. Tetrahedron 1999, 55, 14535-14546. [ Links ] (e) Aguilar, R.; Reyes, A.; Orduña, A.; Zepeda, G.; Bates, R. W.; Tamariz, J. Rev. Soc. Quím. Méx. 2000, 44, 91-96. [ Links ]
5. Herrera, R.; Jiménez-Vázquez, H. A.; Modelli, A.; Jones, D.; Söderberg, B. C.; Tamariz, J. Eur. J. Org. Chem. 2001, 4657-4669. [ Links ]
6. Peralta, J.; Bullock, J. P.; Bates, R. W.; Bott, S.; Zepeda, G.; Tamariz, J. Tetrahedron 1995, 51, 3979-3996. [ Links ]
7. (a) Rappoport, Z., Ed., The Chemistry of Enamines. Wiley, Chichester, 1994. [ Links ] (b) Greenhill, J. V. Chem. Soc. Rev. 1977, 6, 277-294. [ Links ] (c) Granik, V. G. Russ. Chem. Rev. 1984, 53, 383-404. [ Links ]
8. (a) Scott, K. R.; Edafiogho, I. O.; Richardson, E. L.; Farrar, V. A.; Moore, J. A.; Tietz, E. I.; Hinko, C. N.; Chang, H.; El-Assadi, A.; Nicholson, J. M. J. Med. Chem. 1993, 36, 1947-1955. [ Links ] (b) Kesten, S. J.; Degnan, M. J.; Hung, J.; McNamara, D. J.; Ortwine, D. F.; Uhlendorf, S. E.; Werbel, L. M. J. Med. Chem. 1992, 35, 3429-3447. [ Links ]
9. For some recent examples: (a) Bhathia, G. S.; Lowe, R. F.; Pritchard, R. G.; Stoodley, R. J. Chem. Commun. 1997, 1981-1982. [ Links ] (b) Abbiati, G.; Clerici, F.; Gelmi, M. L.; Gambini, A.; Pilati, T. J. Org. Chem. 2001, 66, 6299-6304. [ Links ] (c) Clerici, F.; Gelmi, M. L.; Pocar, D.; Pilati, T. Tetrahedron: Asymmetry 2001, 12, 2663-2669. [ Links ] (d) Buñuel, E.; Gil, A. M.; Díaz-de-Villegas, M. D.; Cativiela, C. Tetrahedron 2001, 57, 6417-6427. [ Links ] (e) Clerici, F.; Gelmi, M. L.; Gambini, A.; Nava, D. Tetrahedron 2001, 57, 6429-6438. [ Links ] (f) Maekawa, K.; Igarashi, T.; Kubo, K.; Sakurai, T. Tetrahedron 2001, 57, 5515-5526. [ Links ] (g) Caine, D. Tetrahedron 2001, 57, 2643-2684. [ Links ] (h) Stanovnik, B.; Svete, J. Synlett 2000, 1077-1091. [ Links ] (i) Yonehara, K.; Ohe, K.; Uemura, S. J. Org. Chem. 1999, 64, 9381-9385. [ Links ]
10. (a) Sustmann, R. Tetrahedron Lett. 1971, 2721-2724. [ Links ] (b) Houk, K. N.; Sims, J.; Watts, C. R.; Luskus, L. J. J. Am. Chem. Soc. 1973, 95, 7301-7315. [ Links ]
11. Jiménez-Vázquez, H. A.; Ochoa, M. E.; Zepeda, G.; Modelli, A.; Jones, D.; Mendoza, J. A.; Tamariz, J. J. Phys. Chem. A 1997, 101, 10082-10089. [ Links ]
12. (a) Guseinov, F. I.; Yudina, N. A.; Burangulova, R. N.; Ryzhikova, T. Ya.; Valliullina, R. Zh. Chem. Heterocyclic Comp. 2002, 38, 496-497. [ Links ] (b) Toplak, R.; Svete, J.; Stanovnik, B.; Grdadolnik, S. G. J. Heterocyclic Chem. 1999, 36, 225-235. [ Links ] (c) Pizzioli, L.; Ornik, B.; Svete, J.; Stanovnik, B. Helv. Chim. Acta 1998, 81, 231-235. [ Links ]
13. Zhuo, J.-C. Magn. Reson. Chem. 1998, 36, 565-572. [ Links ]
14. (a) Fleming, I. Frontier Orbitals and Organic Chemical Reactions. John Wiley & Sons, Chichester, 1976. [ Links ] (b) Houk, K. N. Acc. Chem. Res. 1975, 8, 361-369. [ Links ] (c) Eisenstein, O.; Lefour, J. M.; Anh, N. T.; Hudson, R. F. Tetrahedron 1977, 33, 523-531. [ Links ] (d) Spíno, C.; Pesant, M. Dory, Y. Angew. Chem., Int. Ed. 1998, 37, 3262-3265. [ Links ]
15. (a) Carruthers, W. Cycloaddition Reactions in Organic Synthesis. Pergamon Press, Oxford, 1990, 269-331. [ Links ] (b) Houk, K. N.; Chang, Y.-M.; Strozier, R. W.; Caramella, P. Heterocycles 1977, 7, 793-799. [ Links ] (c) Huisgen, R., in: 1,3-Dipolar Cycloaddition Chemistry, Vol. 1, Padwa, A., Ed., Wiley-Interscience, New York, 1984. [ Links ] (d) Jarosková, L.; Fisera, L.; Matejková, I.; Ertl, P.; Prónayová, N. Monatsh. Chem. 1994, 125, 1413-1425. [ Links ] (e) Houk, K. N.; Sims, J.; Duke, Jr., R. E.; Strozier, R. W.; George, J. K. J. Am. Chem. Soc. 1973, 95, 7287-7301. [ Links ]
16. (a) Sauer, J.; Sustmann, R. Angew. Chem., Int. Ed. Engl. 1980, 19, 779-807. [ Links ] (b) Anh, N. T.; Canadell, E.; Eisenstein, O. Tetrahedron 1978, 34, 2283-2288. [ Links ]
17. (a) Exner, O. J. Phys. Org. Chem. 1999, 12, 265-274. [ Links ] (b) Charton, M. J. Phys. Org. Chem. 1999, 12, 275-282. [ Links ] (c) Galkin, V. I. J. Phys. Org. Chem. 1999, 12, 283-288. [ Links ]
18. Zhuo, J.-C.; Schenk, K. Helv. Chim. Acta 1997, 80, 2137-2147. [ Links ]
19. Allen, F. H.; Kennard, E.; Watson, D. G.; Brammer, L.; Orpen, A. G.; Taylor, R. J. Chem. Soc., Perkin Trans. 2 1987, S1-S19. [ Links ]
20. (a) Fox, M. A.; Cardona, R.; Kiwiet, N. J. J. Org. Chem. 1987, 52, 1469-1474. [ Links ] (b) Bachler, V.; Mark, F. Theoret. Chim. Acta 1976, 43, 121-135. [ Links ] (c) Tripathy, R.; Franck, R. W.; Onan, K. D. J. Am. Chem. Soc. 1988, 110, 3257-3262. [ Links ] (d) Padwa, A.; Kline, D. N.; Koehler, K. F.; Matzinger, M.; Venkatramanan, M. K. J. Org. Chem. 1987, 52, 3909-3917. [ Links ]
21. Kahn, S. D.; Pau, C. F.; Overman, L. E.; Hehre, W. J. J. Am. Chem. Soc. 1986, 108, 7381-7396. [ Links ]
22. The authors have deposited the atomic coordinates for this structure with the Cambridge Crystallographic Data Centre. The coordinates can be obtained, on request, from the Director Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge, CB2 1EZ, UK.
23. SHELXTL, v. 5.03, Siemens Energy & Automation, Germany, 1995. [ Links ]
24. Calculated with Gaussian 94, Revision E.2: Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Gill, P. M. W.; Johnson, B. G.; Robb, M. A.; Cheeseman, J. R.; Keith, T.; Petersson, G. A.; Montgomery, J. A.; Raghavachari, K.; Al-Laham, M. A.; Zakrzewski, V. G.; Ortiz, J. V.; Foresman, J. B.; Cioslowski, J.; Stefanov, B. B.; Nanayakkara, A.; Challacombe, M.; Peng, C. Y.; Ayala, P. Y.; Chen, W.; Wong, M. W.; Andres, J. L.; Replogle, E. S.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Binkley, J. S.; Defrees, D. J.; Baker, J.; Stewart, J. P.; Head-Gordon, M.; Gonzalez, C.; Pople, J. A. Gaussian, Inc., Pittsburgh, PA, 1995; [ Links ] and Mac-Spartan, v. 1.0, WaveFunction Inc., 18401 VonKarman, Suite 370, Irvine, CA 92715. [ Links ]
25. Dewar, M. J. S.; Zoebisch, E. G.; Healy, E. F.; Stewart, J. J. P. J. Am. Chem. Soc. 1985, 107, 3902-3909. [ Links ]

