










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de la Sociedad Química de México
Print version ISSN 0583-7693
Rev. Soc. Quím. Méx vol.46 n.1 Ciudad de México Jan./Mar. 2002
Investigación
Cálculo de la estructura de pequeños cúmulos de moléculas de agua usando un potencial empírico
Eduardo González-Jiménez,1* Guillermina Cedillo-del Rosario,1 Alexandra Deriabina,1 Alexander V. Teplukhin2 y Valery I. Poltev1,3
1 Fac. Ciencias Físico-Matemáticas, (FCFM-BUAP) Ciudad Universitaria 18 sur y Av. San Claudio, Col. San Manuel, Puebla 72570, Pue. E-mail: gonzalez@fcfm.buap.mx
2 Instituto de Problemas Matemáticos de Biología, Academia de Ciencias de Rusia, Pushchino, Región de Moscú, 142290 Rusia.
3 Instituto Teórico y Experimental de Biofísica, Academia de Ciencias de Rusia, Pushchino, Región de Moscú, 142290 Rusia.
Recibido: 24 de mayo del 2001.
Aceptado: 30 de noviembre del 2001.
Resumen
Con el método de Monte Carlo y utilizando un potencial empírico recién ajustado se calculó la estructura de los mínimos locales de energía de interacción de cúmulos de moléculas de agua (H2O)n con n = 2-8. Se obtiene una relación aproximadamente lineal del cúmulo con la energía de interacción. Obtenemos que las estructuras cíclicas son las más estables para n = 3-5. Para cúmulos con seis moléculas de agua la configuración más estable corresponde a la estructura de "libro abierto". Es una estructura compacta para cúmulos con 7 y 8 moléculas de agua, conteniendo estructuras regulares cuadradas. Existe una correlación entre las distancias entre oxígenos del cúmulo con la medida del cúmulo.
Palabras clave: Cúmulos, molécula de agua, funciones de potencial empíricas, método de Monte Carlo.
Abstract
The interaction energy and structure of local minima of water clusters (H2O)n with n = 2-8 has been calculated using Monte Carlo method and recently refined empirical potential functions. A nearly lineal relation between the interaction energy and the cluster size was found. We have found, that cyclic structures are the most stable ones of n = 3-5. For the clusters of six water molecules the most stable configuration corresponds to "open book" sutructure. The configurations of clusters of 7 and 8 water molecules are compact ones and contain nearly regular quadrangles. The correlation exists between inter-oxygen distances and the cluster size.
Keywords: Clusters, water molecules, empirical potential function, Monte Carlo method.
Introducción
El comportamiento de las moléculas de agua ha sido extensivamente investigado desde el punto de vista teórico conduciendo a la construcción y refinamiento de diferentes modelos que describen parcial- y complementariamente muchas de las propiedades del agua líquida [1-3] y cúmulos de moléculas de agua [4-8].
Algunos de los modelos propuestos tienen el interés de estudiar sistemas biológicos, por ejemplo en sistemas que consideran la hidratación de la doble hélice del ADN, se incluye la interacción entre moléculas de agua y ellas con las biomoléculas [9]. Por ejemplo la más conocida es la formación de una doble capa de moléculas de agua o "espina de hidratación" en el surco menor de la doble espiral del ADN-B [10] y pentágonos de hidratación en el octámero de la forma A del ADN [11].
Los modelos de interacción agua-agua son usualmente probados para ver que tan bien describen las propiedades del "bulk" de agua, ya que experimentalmente se dispone de datos suficientes para describir la estructura del agua [12].
Pero en los sistemas biológicos el agua se encuentra en un amplio rango de ambientes, lejos del ADN el agua tiene una estructura similar al agua líquida pero cerca del ADN la estructura puede ser muy diferente, en promedio las moléculas de agua estarán menos involucradas en la formación de enlaces de hidrogeno que en el "bulk" de agua y estarán menos propensas a fluctuaciones, en este sentido uno tiene una situación análoga a la de un cúmulo [10, 11].
Por lo tanto un detallado estudio de los cúmulos de agua contribuye a entender, no solo un amplio rango de problemas químicos que incluyen la estructura de la fase condensada, la solvatación, y la naturaleza del enlace de hidrógeno sino que en muchas áreas de la bioquímica y biofísica involucra procesos en los seres vivos.
El estudio de los cúmulos por otro lado llena el vacío entre nuestro conocimiento de las moléculas y el estado macroscópico de la materia, los cuales son descritos por la mecánica estadística.
En los últimos años un creciente interés en el estudio teórico de cúmulos ha sido estimulado por el aumento en la potencia de cálculo de las modernas computadoras, además que para el estudio de estos sistemas no se recurre a artificios como son: el truncamiento del potencial o las condiciones periódicas a la frontera, siendo por tanto, los cúmulos un objeto ideal para la simulación computacional.
Metodología de Cálculo
Modelo de interacción agua-agua
Los cálculos reportados en este estudio son efectuados con un campo de fuerza que considera las interacciones entre moléculas de agua, dentro de un esquema para simular las interacciones en sistemas con ácidos nucleicos y solvente acuoso. El potencial fue ajustado en 1984 [9] y optimizado recientemente a los nuevos resutados experimentales [13].
Las funciones usadas son potenciales par efectivos para monómeros de moléculas de aguas rígidas de tres puntos. Las cargas en los átomos de la molécula de agua son +0.34 y -0.68 (partes de la carga del electrón) para los átomos de hidrógeno y oxígeno respectivamente, estos valores dan una magnitud de 1.9 D para el momento dipolar del agua, este valor es cercano al valor experimental (1.855D [14]). Los demás parámetros geométricos utilizados en la molécula de agua son, la longitud del enlace O-H de 0.98 Å y un ángulo H-O-H tetraédrico 109.47°.
Las expresiones algebraicas de las funciones de potencial tienen una forma simple, constan de una interacción coulombiana entre todas las cargas puntuales por pares de átomos intermoleculares y términos no-coulombianos para describir la interacción entre átomos de oxígeno y de hidrógeno que permiten formas enlaces de hidrógeno:
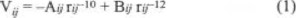
con los valores de A y B de 7193 y 18356, respectivamente. Además términos de Lennard-Jones para interacciones entre átomos de oxígeno o entre átomos de hidrógeno:
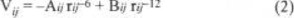
con coeficientes para OO de 366 y 882847, y para HH de 35 y 5914 para A y B respectivamente. El ajuste de los coeficientes se hizo para reproducir las características energéticas y estructurales del agua líquida, obteniendo para la energía de interacción intermolecular a 300 K de -10.04 kcal / mol por par molecular [13] (el valor experimental es de -9.92 kcal / mol [1, 2]).
Procedimiento de simulación
Para la obtención de las estructuras en los mínimos locales de cúmulos de moléculas de agua (H2O)n con n = 2-8, se realiza, con ayuda del método de Monte Carlo, algoritmo de Metrópolis el cual fue descrito en otros trabajos [9, 15], estas estructuras se encuentran cuando se baja la temperatura del sistema durante los cálculos en el proceso de Monte Carlo, este tipo de estructuras son llamadas estructura F [16]. En calidad de configuración inicial se construyen estructura I o estructuras instantáneas que resultan de la simulación de Monte Carlo a 300 K.
Las estructuras iniciales corresponden a mínimos de energía reportadas por otros autores [4-7, 17, 18]. Partiendo de estas configuraciones iniciales procedemos a calcular las estructuras F a una temperatura cercana al cero absoluto (0.5 K). Las moléculas de agua durante el procedimiento de Monte Carlo son desplazadas y rotadas con valores máximos de 0.012 Å y 0.012 rad., respectivamente. Los mínimos locales se obtuvieron cuando los valores de la energía no presentan variaciones, en promedio se realizan 105 pasos de Monte Carlo por molécula de agua por estructura. Para valorar la formación de enlaces de hidrógeno entre las moléculas de agua utilizamos un criterio geométrico sobre el criterio energético [3].
Resultados y discusión
En la tabla 1 presentamos las características energéticas y la estructura de algunos de los cúmulos de moléculas de agua (H2O)n calculados para n = 2-8. Para cada n se obtienen un conjunto de mínimos locales. De los cálculos realizados se encuentra una correlación directa entre la medida del cúmulo con la energía de interacción (Fig. 1) el ajuste de los datos muestran una correlación aproximada lineal, la dependencia de E(n) da un aumento en valor absoluto de 9.83 kcal / mol por cada molécula que se adiciona al cúmulo. En la Tabla 1 presentamos también el número de enlaces por cada molécula del cúmulo y de estos enlaces cuántos de ellos son donores y cuántos aceptores. En la Fig. 2 mostramos los cúmulos más energéticos obtenidos de nuestra simulación.

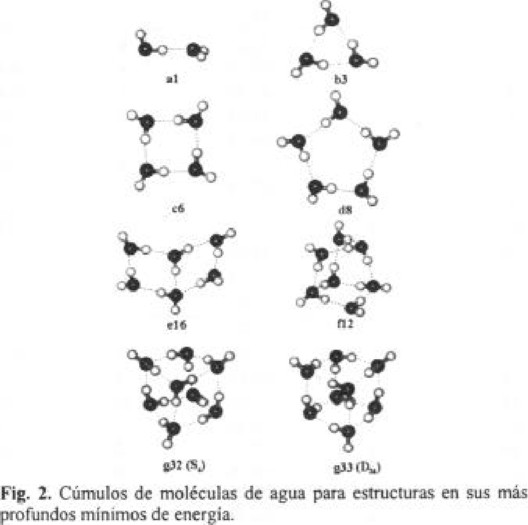
Una de las estructuras más estudiadas y para la cual existen un gran número de trabajos teóricos [4 y referencias en él] y mediciones experimentales [12, 19-22] es el dímero. Nuestros resultados para la geometría óptima del dímero de agua corresponden a una estructura con simetría plana en configuración trans y un enlace de hidrógeno lineal (a1), de acuerdo a resultados experimentales de espectroscopía de microondas la distancia entre oxígenos en la molécula, R(OO) es de 2.98 ± 0.04 Å [14] y con correcciones inarmónicas se estima de 2.946 Å [21] con un ángulo OHO de 6 ± 20° [14].
Los valores calculados por nosotros para la estructura F (0.5K) del dímero son de 2.804 Å y 2.9 ° respectivamente para la longitud y el ángulo del enlace, el valor de la energía es de -6.48 kcal / mol. Considerando que estos valores fueron calculados cercanos al cero absoluto y para efectos de comparación realizamos una simulación a 300 K. Durante la simulación las moléculas de agua se evaporan en promedio después de 7 × 103 pasos de Monte Carlo. Los resultados reportan que la distancia R(OO) varía de 2.85Å a 2.99Å y el ángulo OHO es de 5.6° para el dimero. La energía de interacción resultó ser de -4.52 kcal / mol que es cercano al valor experimental de entalpía de formación de -4.2 ± 1.1 kcal / mol, obtenido por espectroscopia de masas en vacío a 298K [22] y al valor de la energía de interacción calculado de mediciones de conductividad térmica por Curtis y colaboradores [20] que reportan la cantidad de -5.44 ± 0.7 kcal / mol. El valor calculado con el potencial ajustado y temperatura cercana al cero absoluto es de -6.48 kcal / mol.
De nuestros resultados se puede ver la tendencia del acortamiento del enlace de hidrógeno al congelar el dímero, esto se refleja en las distancias R(OO) cuyos valores van de 2.99 Å a 2.804 Å, el momento dipolar calculado a 0.5 K es de 2.99 D, un poco mayor al experimental (2.601 ± 0.005 D, [19]).
En la Tabla 2 mostramos las características energéticas de los más profundos mínimos para los cúmulos de n = 2-8 calculados y obtenidos experimentalmente [5, 17, 18, 22, 23] estos corresponden a la Fig. 2.
La discrepancia de nuestros resultados de las energías calculadas respecto a los datos experimentales se debe principalmente a que en el ajuste de los parámetros para las funciones de potencial para la interacción agua-agua se hizo para el "bulk" de agua [13], esto implica que toma en cuenta la no-aditividad de las interacciones, producidas principalmente por las contribuciones de polarización. Por ello para cúmulos pequeños la coincidencia no es tan buena cuantitativamente como se observa en la Tabla 2. y en el dimero a bajas temperaturas.
Para las estructuras n = 3-5 los mínimos corresponden a estructuras cíclicas homodrómicas con hidrógenos enlazados en una dirección y los átomos de oxígeno cuasi planares.
Observaciones experimentales recientes para el trímero, tetrámero y pentámero de moléculas de agua con espectroscopía FIR-VRT [5] confirman que la estructura en equilibrio corresponde a estructuras similares a las encontradas por nosotros como también es predicho por numerosos cálculos cuánticos [4, 6, 24].
La estructura del trímero y tetrámero de agua es un anillo con sus enlaces O-H libres en el plano a diferencia de la estructura reportada en la literatura [6-8] con sus hidrógenos libres hacia un lado del plano del anillo (b3 y c6). Esto puede ser consecuencia a que las distancias entre los átomos de hidrógeno libres en estas estructuras se encuentran más alejados (4.4 Å y 4.1 Å para el trímero y tetrámero) no así para las estructuras cíclicas con n > 4 (3.9 Å para el pentámero con distorsión) pues las repulsiones son considerables y suceden plegamiento de las estructuras.
Las distancias R(OO) calculadas por nosotros varían de 2.823 Å a 2.833 Å, parecidas a las reportadas con mecánica cuántica: en cálculos de perturbaciones MP2 varían de 2.798 Å a 2.800 Å [6], y de 2.825 Å a 2.837 Å con métodos de cúmulos acoplados [7] y de 2.780 Å a 2.784 Å utilizando métodos de funcionales de la densidad [4] todos los cálculos presentan una discrepancia con los valores experimentales [23, 25] que reportan de 2.94 Å a 2.97 Å, la discrepancia puede explicarse debido a que esta estructura experimental se obtuvo de un reducido conjunto de constantes rotacionales pero estas no son únicas.
Para la estructura del tetrámero cíclico calculado con nuestro potencial, la longitud R(OO) es menor que el trímero, el valor de 2.799 Å a 2.808 Å y con valores de 2.797 Å a 2.799 Å para el pentámero. Esta tendencia de disminuir la distancia entre los átomos OO al incrementar el número de cúmulos se observa también en [4, 5]. Sobre mediciones de constantes rotacionales en cúmulos [5] las distancias OO tienden a acortarse, estos datos experimentales indican una rápida convergencia al valor del "bulk" ordenado de agua al incrementar la medida del cúmulo (R(OO) para el hielo Ih es de 2.759 Å a 223 K [26] y 2.975 Å para agua líquida a 298.2 K [12]). La dependencia del acortamiento de las distancias OO obtenida es similar a la del trabajo [18] que reporta una relación exponencial para la contracción y para el pentámero valores de 2.711 Å a 2.717 Å. Esta aparente contracción muestra los efectos cooperativos en la red de enlaces de hidrógeno.
La estructura en el equilibrio del pentámero como ya mencionamos es cíclica pero contraída, la estructura es similar a la forma inclinada del ciclopentano. Cada molécula actúa como un donador y un aceptor de enlaces de hidrógeno. En este cúmulo (H2O)5 los hidrógenos libres están desplazados arriba y abajo del plano en contraste con la geometría cuasi planar para n = 3 y 4. La estructura pentagonal es asimétrica, tres átomos de oxígeno están aproximadamente en el plano y los restantes a 4 ° y 18 ° fuera del plano (d8). En general para cúmulos cíclicos (HsO)n con n impar (para n ≥ 5) el patrón de las estructuras requiere que dos hidrógenos vecinos adyacentes estén hacia el mismo lado resultando una estorbo estérico siendo ésta la principal causa de la distorsión del cúmulo respecto al plano, en cambio para n par la estructura es más simétrica y hay menos distorsión, esto se puede ver en los momentos dipolares calculados para todas las estructuras cíclicas con valores de 0.234 D y 0.188 D para n = 5 y 7, y los valores 0.017 D y 0.083 D para n = 6 y 8. Los valores del momento dipolar de las estructuras cíclicas con n = 3, 4, 5 son muy próximos a los valores calculados y medidos experimentalmente [27, 28].
Respecto al hexámero cálculos ab initio [29] de niveles superiores predicen estructuras 3D compactas como las más estables. Otros estudios mecánico cuánticos en el nivel de Hartree-Fock encuentran que las estructuras cíclicas y de libro abierto tienen mínimos globales casi isoenergéticos [30]. Cálculos con teoría de la densidad funcional (DFT), predicen que la estructura cíclica es el mínimo global [31] esto es reafirmado en [32]. Kim y col. reportan a la estructura "cage" como la mas energética [33]. De lo anterior notamos que el hexámero se sitúa en la región intermedia entre las estructuras cíclicas y compactas.
De nuestros cálculos, la estructura de libro (e16) resultó ser la más energética con una diferencia de 1.7 kcal / mol respecto al cúmulo cíclico (e13). Pero con una diferencia de 0.39 kcal / mol respecto a la estructura de "cage" (e15) y una diferencia mayor para el prisma (e9) cuyo valor es de 42.3 kcal / mol. En [33] la diferencia entre las estructuras "cage" y el libro es de 0.01 kcal / mol.
El momento dipolar de la estructura de libro abierto es de 1.985 D a diferencia de la cíclica que es no polar. Nuestro resultado es mas acorde al valor experimental de 1.85 D [34] mientras que de los resultados ab initio la estructura del libro es de 2.487 D [33] y 2.77 D en [35]. El rango de distancias para nuestro mínimo va de 2.795 Å a 2.832 Å.
Para los cúmulos con n < 6 se observa que las estructuras con más enlaces de hidrógeno son menos estables, esto es la conformación cíclica es más estable que otros conformeros estos tienen un momento dipolar casi nulo (Tabla 2), mientras que las otras estructuras en su mínimo tienen un significativo momento dipolar (mayor o cercano a 2D). Resultados ab initio parecen estar de acuerdo con nosotros y los experimentos los cuales no hallan significativos momentos dipolares en cúmulos de agua con n < 6 excepto para el dímero [14]. Para estructuras con n = 6,7,8 las más energéticas tienen mas enlaces-H, y considerables momentos dipolares. El heptámero con un momento dipolar comparable al obtenido de cálculos ab initio [36] como se ve en la Tabla 2.
En la figura 3 graficamos la energía de interacción por molécula de agua de los más profundos mínimos contra la medida del cúmulo es fácil notar una correlación la cual se puede expresar en una función simple

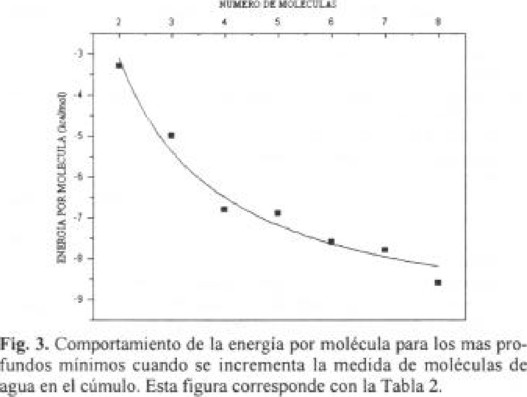
Un ajuste de la curva a los datos, se obtiene en las constantes A y B, tiene los valores de -9.90 y 13.57 respectivamente. Cálculos mecánicos cuánticos DFT [24] reportan resultados cualitativamente similares con constantes correspondientes -12.04 y 18.45 pero sus datos están más dispersos ya que las desviaciones son menores para nuestros resultados. De la figura 3 se observa para n = 4,8 los cúmulos son relativamente más estables que el resto, nuestros resultados refuerzan la suposición de Lee [24] que los sistemas de moléculas de agua más estables se forman maximizando el número de estructuras planas con anillos de cuatro miembros. Observamos (Tabla 1, Fig. 4) que en estructuras para n = 7 las más energéticas son una estructura compacta f12 y una abierta f11 con dos formas cuadradas homodrómicas. Para 8 moléculas de agua la estructura más estable es el cubo, en la figura 2 presentamos dos cúmulos cúbicos (H2O)8 con simetrías D2d y S4 aproximadamente isoenergéticos (g32 y g33) con predominio de la estructura de simetría D2d como es reportado por otros autores [35], la diferencia es de 0.4 kcal / mol. Esta estructura da el mayor número de formas cuadradas (6), pero aún las próximas a estas, las g27 y g25 contienen 3 formas cuadradas. Otra estructura interesante es la g24 abierta con tres formas cuadradas, esta es la mínima de un conjunto de estructuras similares con energías que van de -57.9 a -70.3 kcal / mol de esta serie de estructuras se ve la disposición a cerrar la estructura, ésta parece verse favorecida cuando las estructuras son homodrómicas. La tendencia a las formas cuadradas homodrómicas se observa ya en el hexámero cuya estructura de mínima energía es el libro (e16) con 1.89 kcal / mol más que la estructura cíclica (e13). Estas estructuras tienen caras exteriores hacia donde los hidrógenos libres se dirigen, a diferencia del "bulk" de agua que presenta predominancia de otras estructuras, como en hielo normal (Ih) cuya estructura consiste de arreglos regulares de hexágonos, o de cálculos de dinámica molecular para agua líquida (298.2 K) reportan una estructura dominada por pentágonos [37].

Conclusiones
La energía de interacción de los cúmulos respecto a su medida muestra una correlación lineal con el aumento de 9.92 kcal / mol por cada molécula que se adiciona al cúmulo. Los mínimos más profundos para cúmulos con 3, 4 y 5 moléculas de agua son estructuras cíclicas homodrómicas, tal como lo reportan otros autores. Para 6 moléculas de agua el mínimo corresponde a la estructura de "libro abierto" con 1.6 kcal / mol mayor que la estructura cíclica, este conformero es la transición a estructuras compactas más estables como la cúbica. Se observa una dependencia hiperbólica de la energía por molécula con la medida del cúmulo, se observa una predominancia de estructuras cuadradas en los cúmulos. Un cálculo para cúmulos con mayor número de moléculas de agua (que esta en proceso) reafirmaría la última proposición.
Agradecimientos
Este trabajo fue financiado por CONACyT a través del proyecto 35239-E.
Referencias
1. Eisenberg, D.; Kauzmann, W. The structure and Properties of Water, Oxford University Press. London, 1969. [ Links ]
2. Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. J. Chem. Phys. 1983, 79, 926-935. [ Links ]
3. Malenkov, G.G.; Tytik, D.L.; Zheligovskaya, E.A. J. Mol. Liquids, 1999, 82, 27-38. [ Links ]
4. Estrin, D.A.; Paglieri L.; Corongiu, G.; Clementi, E. J. Phys. Chem. 1996, 100, 8701-8711. [ Links ]
5. Liu, K.; Cruzan, M.G.; Saykally, R.J. Science, 1996, 271 929-933. [ Links ]
6. Xantheas, S.S.; Dunning, T.H. Jr. J. Chem. Phys. 1993, 99, 8774-8792. [ Links ]
7. Fowler, J.E.; Schaefer, H.F. J. Am. Chem. Soc. 1995, 117, 446-452. [ Links ]
8. Mo, O.; Tanez, M., Elguero, J. J. Chem. Phys. 1992 97, 6628-6634. [ Links ]
9. Poltev, V.I.; Grokhlina, T.I.; Malenkov G.G. J. Biomol. Struct. Dyn. 1984, 2, N2, 413-429. [ Links ]
10. Drew H.R.; Dickerson R.E. J. Mol. Biol., 1981, 151, 535-556. [ Links ]
11. Kennard O.; Cruse W.B.T.; Nachman J.; Prange T.; Shakked Z.; Rabinovich D., J. Biomol. Struct. Dyn. 1986, 3, 623-647. [ Links ]
12. Soper, A.K.; Phillips M.P. Chemical Physics 1986, 107, 47-60. [ Links ]
13. González, E.; Cedeño, F.I.; Teplukhin, A.V.; Malenkov, G.G.; Poltev, V.I. Rev. Mex. Fis. 2000, 46, S.2, 142-147. [ Links ]
14. Dyke, T.R.; Muenter, J.S. J. Chem. Phys. 1973, 59, 3125-3127. [ Links ]
15. Poltev V.I., Malenkov G.G., Gonzalez E., Teplukhin A.V., Rein R. Shibata M., Miller J.H. J. Biolmol. Struct. Dyn. 1996, 13, N.4, 717-725. [ Links ]
16. Malenkov, G.G. The Chemical Physics of Solvation, Part. A., Amsterdam: Elsevier, 1985, 355-389. [ Links ]
17. Cruzan, J.D.; Braly, L.B.; Liu, K.; Brown, M.G.; Loeser, J.G.; Saykally, R.J. Science 1996, 271, 59-62. [ Links ]
18. Liu, K.; Brown, M.G.; Cruzan, J.D.; Saykally, R.J. Science, 1996, 271, 62-64. [ Links ]
19. Kuchitsu, K.; Morino, Y. Bull. Chem. Soc. Jpn. 1965, 38, 805-810. [ Links ]
20. Curtiss, L.A.; Frurip, D.L.; Blander, D.L. J. Chem. Phys. 1979, 71, 2703-2709. [ Links ]
21. Odutola, J.A.; Dyke, T.R. J. Chem. Phys. 1980, 72, 5062-5070. [ Links ]
22. Sukhodub, L.F. Chem. Rev. 1987, 87, 589-606. [ Links ]
23. Pugliano, N.; Saykally R.J. Science 1992, 257, 1937-1940. [ Links ]
24. Lee C., Chen H., Fitzgerald G. J. Chem. Phys. 1995 102, 1266-1269. [ Links ]
25. Wei, S.; Shi, Z.; Castleman, A.W. J. Chem. Phys. 1991, 94, 3268-3270. [ Links ]
26. Peterson, S.W.; Levy, H.A. Acta Crystallogr. 1957, 10, 70-76. [ Links ]
27. Vernon, M.F.; Krajnovich, D.J.; Kwok, H.S.; Lisy, J.M.; Shen, Y.R.; Lee, Y.T. J. Chem. Phys. 1982, 77, 47-57. [ Links ]
28. Knochenmuss, R.; Leutwyler, S. J. Chem. Phys. 1992, 96, 5233-5244. [ Links ]
29. Kim, K.D.; Jordan, T.S.; Zwier, J. Am. Chem. Soc. 1994, 116, 11568-11569. [ Links ]
30. Mhin, B.J.; Kim, H.S.; Kim, H.S.; Yoon, C.W.; Kim, K.S. Chem. Phys. Lett. 1991, 176, 41-49. [ Links ]
31. Laasonen, K.; Parrinello, M.; Car, R.; Lee, C.; Vanderbilt, D. Chem. Phys. Lett. 1993, 207, 208-211. [ Links ]
32. Mhin,B.J.; Kim, J.; Lee, S.; Lee, J.Y.; Kim, K.S. J. Chem. Phys. 1994, 100, 4484-4486. [ Links ]
33. Kim J.; Kim K.S., J. Chem. Phys., 1998, 109, 5886-5895. [ Links ]
34. Liu K.; Brown M.G.; Carter C.; Saykally R.J.; Gregory J.K.; Clary D.C. Nature (London) 1996, 381, 501-503. [ Links ]
35. Day P.N.; Pachter R.; Gordon M.S.; Merrill G.N. J. Chem. Phys. 2000, 112, 2063-2073. [ Links ]
36. Kim J.; Majumdar D.; Lee H.M.; Kim K.S. J. Chem. Phys. 1999, 110, 9128-9134 [ Links ]
37. Rahman, A.; Stillinger F.H. J. Am. Chem. Soc. 1973, 95, 7943-7951. [ Links ]

