



![Síntesis y estudio estructural por RMN de ¹H y 13C de la N-[4-[2-(2-oxo-2H-1-benzopiranil-3-carboxamidil)etil]bencensulfonil]-N'-ciclohexilurea y de la N-[4-[2-(4-nitrobenzamidil)etil]-bencensulfonil]-N'-ciclohexilurea](/img/en/prev.gif)






Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de la Sociedad Química de México
Print version ISSN 0583-7693
Rev. Soc. Quím. Méx vol.45 n.4 Ciudad de México Oct./Dec. 2001
Revisión
Enthalpic and Entropic Contributions to the Conformational Free Energy Differences in Monosubstituted Cyclohexanes
Eusebio Juaristi* and Omar Muñoz-Muñiz
Departamento de Química, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, Apartado Postal 14-740, México 07000, D. F. E-mail: juaristi@relaq.mx
Recibido el 10 de febrero del 2001.
Aceptado el 16 de julio del 2001.
Dedicated to Professor Fernando Walls, Instituto de Química, UNAM, on the occasion of his 70th Birthday.
Abstract
Variable-temperature 1H and 13C NMR spectroscopy of substituted cyclohexanes permitted the evaluation of the thermodynamic parameters for the axial ⇌ equatorial conformational equilibria when the substituent is methyl, ethyl, isopropyl, tert-butyl, benzyl, and the sulfur-containing methylthio, methylsulfinyl, and methylsulfonyl. Interpretation of the results confirms the premise that a proper understanding of conformational preferences requires the knowledge of the enthalpic and entropic contributions to the conformational free energy differences. A comment on the determination of thermodynamic parameters by means of theoretical methods is also included.
Keywords: Conformational analysis, A-values, NMR spectroscopy, Variable-temperature NMR.
Resumen
Estudios espectroscópicos mediante resonancia magnética nuclear a diferentes temperaturas hicieron posible la evaluación de los parámetros termodinámicos asociados a equilibrios conformacionales axial ⇌ ecuatorial en ciclohexanos sustituidos, en los que el sustituyente es metilo, etilo, isopropilo, terbutilo, bencilo, o algunos de los grupos tiometilo, metilsulfinilo o metilsulfonilo. La interpretación de los resultados obtenidos muestra que para alcanzar el entendimiento correcto de las preferencias conformacionales es necesario conocer las componentes entálpicas y entrópicas además de las diferencias en la energía libre conformacional. Finalmente, se presenta también el uso de métodos teóricos para la determinación de parámetros termodinámicos.
Palabras clave: Análisis conformacional, valores-A, Resonancia magnética nuclear, RMN a temperatura variable.
Enthalpic and entropic contributions to the conformational preference of the benzyl group in cyclohexane
Substituted cyclohexanes generally exist in a conformational equilibrium that involves ring inversion, and therefore interconversion of the axial and equatorial orientation of the substituent (Eq. 1).

Inversion of the cyclohexane ring is slow at low temperatures (e.g. −70 °C or lower), so that a NMR spectrum registers then the signals that correspond to each isomer. Integration of the signals allows then determination of the equatorial/axial ratio, i.e. the equilibrium constant K in equation 1. Now, application of Gibbs' equation (Eq. 2) affords the free energy difference for the axial  equatorial equilibrium, which corresponds to the conformational preference (A-value) of the substituent R, where R is the gas constant (1.987 kcal/mol) and T is the temperature (in kelvin degrees) [1].
equatorial equilibrium, which corresponds to the conformational preference (A-value) of the substituent R, where R is the gas constant (1.987 kcal/mol) and T is the temperature (in kelvin degrees) [1].

A-values (differences in free energy, −ΔG°, between the axial and equatorial conformations of monosubstituted cyclohexanes) are of great interest to chemists since they serve as models in the understanding of the conformational behavior of more complex molecules. For example, when R is an alkyl group, the equilibrium shown in equation 1 is displaced to the right since an equatorial orientation of the substituent avoids the repulsive steric interactions with the axial hydrogens at carbons 3 and 5 (Eq. 3). Thus, it is not surprising that the bigger the substituent R, the greater the preference for the equatorial conformation.

However, we must remember that the free energy of a molecule in a particular conformation is the result of two contributing factors: its enthalpy (H°) and its entropy (S°) components, as described in equation 4.

Specifically, the A-values for the methyl, ethyl, and isopropyl groups are 1.74, 1.80 and 2.21 kcal/mol [1], that appear to be congruent with their relative size; that is, isopropyl larger than ethyl, and ethyl larger than methyl. Nevertheless, an NMR study showed that enthalpy differences for this series decreases, contrary to intuition: − ΔH° (CH3) = 1.75 kcal/mol ; − ΔH° (CH3CH2) = 1.60 kcal/mol, and −ΔH° ((CH3)2CH) = 1.52 kcal/mol [2].
The contrasting trends for ΔG° and ΔH° values in the methyl-ethyl-isopropyl series is explained in terms of the entropy differences, which make a substantial contribution to the corresponding free energy differences (Table 1).

We deemed it of interest to carry out the conformational study, by means of NMR spectroscopy, of benzylcyclohexane (Eq. 5). Consideration of the repulsive steric interactions present in the axial and equatorial conformers, leads to the conclusion that the enthalpy difference in the axial to equatorial benzylcyclohexane equilibrium must be lower than the one for methylcyclohexane (Eq. 6), since one of the rotamers in equatorial benzylcyclohexane suffers from increased steric repulsion relative to equatorial methylcyclohexane (Chart 1).
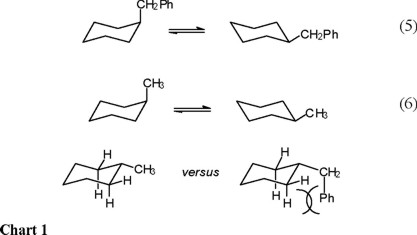
On the other hand, it can be appreciated that the equatorial form of benzylcyclohexane must present three populated rotamers, whereas the axial form only two (the phenyl-inside rotamers, ax-3 in scheme 1, is energetically too unfavorable), so that the ΔS° term (entropy of the axial conformer versus entropy of the equatorial conformer) favors the equatorial benzylcyclohexane.
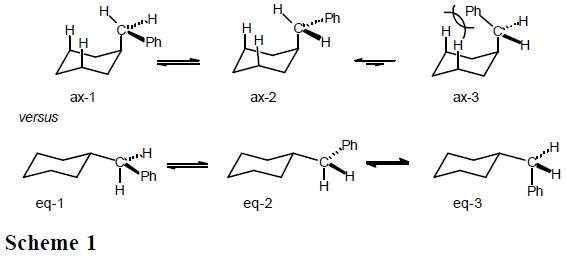
Therefore, the strong steric repulsion between the phenyl and the cyclohexane ring in ax-3 (Scheme 1) brings as consequence that only rotamers ax-1 and ax-2 are populated in the axial conformation. In contrast, it is anticipated that all three equatorial rotamers (ec-1, ec-2, and ec-3 in Scheme 1) are populated, since all three have low energy. In this way, there is more entropy (freedom of movement) in the equatorial conformer; that is, ΔS° = [S°(equatorial) − S°(axial)] > 0.
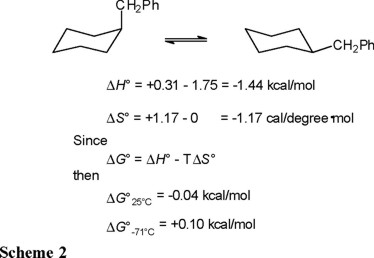
Experimentally [3], the 1H NMR spectrum of cis-1-benzyl-4-methylcyclohexane (cis-1, a cyclohexane derivative where the methyl and benzyl groups compete for the equatorial position) shows a doublet signal with J = 7.9 Hz at δ = 2.57 ppm, which corresponds to the benzylic hydrogens in an averaged spectrum, due to rapid inversion of the cyclohexane ring (Eq. 7) [4].

When the spectrum is registered at −71 °C, the signal for the benzylic hydrogens appears as two doublet signals at 2.65 and 2.46 ppm, in a 56.4:43.6 ratio, an estimated ΔG°202°K = −RT ln K = + 0.10 kcal/mol is obtained. This "doubling" of the spectrum is due to the fact that at low temperature the inversion process is sufficiently slow to permit the recording of the signals associated to cis-1-ax (axial benzyl and equatorial methyl), as well as those corresponding to cis-1-eq; that is, decoalescence of the signals is achieved at −71 °C.
Because the methylene signal in conformationally fixed trans-1 has δ = 2.47 ppm (Eq. 8), a reasonable conclusion is that the downfield signal at δ = 2.65 ppm corresponds to the axial benzyl. Therefore, at low temperature the conformational equilibrium of cis-1 (Eq. 7) is displaced to the left; that is, the conformer with axial benzyl is predominant despite its larger size relative to methyl.

We then recorded 13C NMR spectra for cis-1, both at 25 °C (rapid inversion, averaged spectrum) and at −71 °C (slow inversion, separate spectra for individual conformers). The most relevant signals at ambient temperature are those for the methyl group at δ = 20.40 ppm, and at 41.06 ppm for the benzylic methylene. Below coalescence (at −71 °C), these signals separate into two pairs of signals: one at δ = 17.53 and 44.39 ppm for methyl and benzylic methylene in cis-1-eq, and the second at δ = 23.39 and 37.20 ppm for the same carbons in cis-1-ax. (Eq. 9).

From integration of the C-13 NMR signals for each conformer, a 54.7:45.3 ratio in favor of cis-1-ax was determined (Eq. 9). Again, application of Gibbs' equation affords ΔG°-71°C = −RT ln 45.3/54.7 = +0.08 kcal/mol. It is then confirmed that at low temperature, the conformer with equatorial methyl and axial benzyl predominates, in spite of the larger size of the latter substituent.
Most interestingly, with the results obtained at −71°C, and the application of Eliel's equation [5] (Eq. 10) to room-temperature 13C NMR data gives,

Nevertheless, from Gibbs' equation, ΔG°25°C = −RT ln K = −0.04 kcal/mol. That is, at 25 °C the cis-1-ax  cis-1-eq equilibrium shown in equation 7 favors cis-1-eq, so that now it is the more voluminous substituent that predominates in the equatorial position.
cis-1-eq equilibrium shown in equation 7 favors cis-1-eq, so that now it is the more voluminous substituent that predominates in the equatorial position.
Therefore, the conformational behavior of cis-1 is highly dependent on the experimental temperature of measurement, ΔG°-71°C = + 0.10 kcal/mol versus ΔG°25°C = − 0.04 kcal/mol.
This dependence of ΔG° with temperature shows that entropy plays an important role on the conformational equilibrium of cis-1. Indeed, the setting and solving two equations with two unknowns (ΔH° and ΔS° in equations 11 and 12) [6] allows determination of the thermodynamic parameters (Eqs. 13 and 14).

It is then clear that at low temperature, ΔG° in equation 7 is dominated by the enthalpic term (greater intrinsic preference of the CH3 group to be equatorial), but at ambient or higher temperature (T ≥ 25 °C) ΔG° is dominated by the entropic term TΔS, and as a consequence the observed preferences for the equatorial position follow the "expected" order, PhCH2 > CH3.
The thermodynamic parameters given by equations 13 and 14 refer, of course, to the conformational equilibrium depicted in equation 7. In order to derive the corresponding ΔH° and ΔS° values in benzylcyclohexane (Eq. 5) one must subtract the contributions of the methyl group ("counterpoise" [1]) given in Table 1. In this way, the values presented in Scheme 2 are obtained.
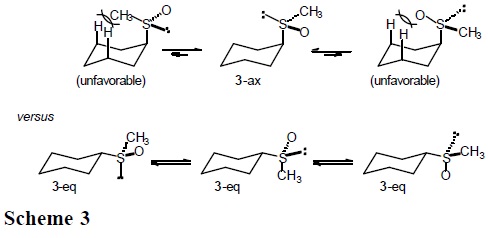
Enthalpic and entropic contributions to the conformational free energies of methylthio, methylsulfinyl, and methylsulfonyl groups in cyclohexane
It is expected that the conformational behavior of cyclohexane derivatives containing the methylthio (CH3S), methylsulfinyl (CH3SO), and methylsulfonyl group (CH3SO2) will depend substantially on the entropic term. In particular, for sulfoxide 3 one anticipates three populated rotamers in the equatorial conformers, but only one in the axial conformation (Scheme 3).
Similarly, three low-energy rotamers are anticipated for the equatorial forms in sulfide 2 and sulfone 4, but only two for the axial conformations (Schemes 4 and 5).
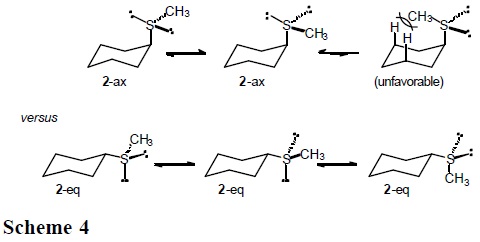

This section reports the results of variable-temperature 13C NMR measurements in (cis-4-methylcyclohexyl)methyl sulfide, sulfoxide and sulfone (5-7 in Scheme 6), which permitted the determination of ΔH° and ΔS° in axial to equatorial equilibria [7]. The cis-methyl at C(4) serves as a counterpoise substituent [1], so that equilibrium constants, K, are closer to unity.

Application of Eliel's equation (Eq. 10) to the C-13 NMR data for 5-7, obtained at various temperatures, afforded the equilibrium constants K that are collected in Table 2. Linear regression analysis of the correlations ln K versus 1/T (Fig. 1) provided the thermodynamic parameters listed in Table 3 [8].
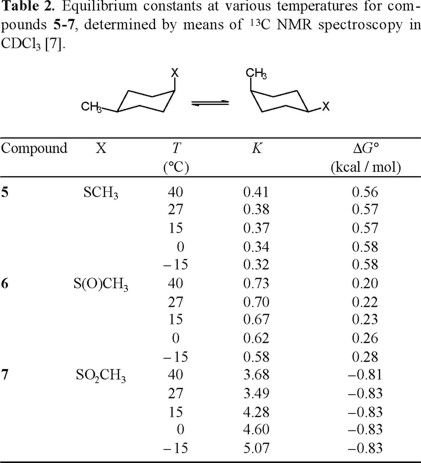


The results reported in Table 3, especially the similarity in enthalpy values for the thiomethyl and methylsulfinyl groups (ΔH° = −1.05 and −1.08 kcal/mol, respectively) are in line with expectation when one considers that for both compounds the axial conformer orients the sulfur lone pair towards the cyclohexane ring, presenting similar steric hindrance. On the other hand, the enthalpy term for the methylsulfonyl group, ΔH°(SO2CH3) = − 2.66 kcal/mol, is more than twice the one for CH3S and CH3S(O), as a consequence of having the sulfonyl oxygen pointing inside the ring (Scheme 5).
The entropy values determined in this work are also congruent in the case of sulfide 2 and sulfoxide 3, since these compounds present three populated rotamers in the equatorial conformation, but only two for axial SCH3 and only one for axial CH3S(O). Indeed, it can be calculated that for X = CH3S, ΔS° = R ln 3 − R ln 2 = +0.80 cal/degree · mol, and ΔS° = R ln 3 − R ln 1 = + 2.18 cal/degree · mol for X = CH3S(O). The experimental values are +0.48 and + 1.55 cal/degree · mol, respectively.
Reinvestigation of the conformational enthalphy, entropy, and free energy of methyl- (8), ethyl- (9), and isopropylcyclohexane (10) [10]
The determination of the conformational energy difference between the axial and equatorial isomers of methylcyclohexane (8) by Booth and Everett (see section A and reference 2) involved measurement of the ratio of the intensities of the enriched 13C resonances in the two isomers 8-ax and 8-eq (Eq. 15).

Wiberg and coworkers [10] have recently questioned the accuracy of the reported equilibrium constants which ranged from K = 164 at 172 K to K = 427 at 149 K, since it was considered that the determination of isomer ratios greater than 100 is extremely difficult. Consequently, the approach used by Wiberg, et al. involved comparision of the NMR signal intensities for natural abundance 13C(2,6) in equatorial methylcyclohexane (δ = 36.1 ppm) versus the small signal for the 13C-enriched axial methyl in 8-ax (δ = 17.6 ppm). In this fashion, K ratios on the order of 4-10 were accurately measured.
Furthermore, Wiberg and coworkers reported the use of a low-temperature "thermometer" based on the temperature-sensitive 13C chemical shifts of 2-chlorobutane, as an internal reference [10,11].
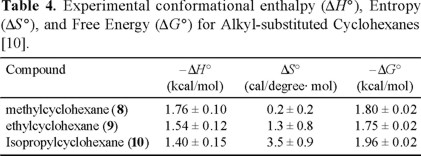
The conformational enthalpy, entropy, and free energy of methyl, ethyl, and isopropyl groups in cyclohexane determined in this study [10] are summarized in Table 4. These values do not differ appreciably from those reported by Booth and Everett [2].
Conformational study of phenylcyclohexane [12]
The conformational free energy (−ΔG° = A-value) of phenylcyclohexane (11) was determined by Eliel and Manoharam from a low-temperature 13C NMR study of cis-4-methyl-1-phenylcyclohexane [13], and a value of ΔG°173 K = −2.87 ± 0.09 kcal/mol was obtained for equation 16.

Very recently, the conformational isomers of phenylcyclohexane (11-ax and 11-eq) were studied via geometry optimization at the HF/6-31G*, B3LYP/6-311G*, and MP2/6-31G* theoretical levels, and the results are summarized in Table 5. At all levels of theory, equatorial phenylcyclohexane (11-eq) was found to preferentially adopt a conformation in which the phenyl is eclipsed with the C(1)-H bond (Fig. 2a). The lowest energy rotational arrangement of the phenyl group in the axial isomer (11-ax) is one in which the plane of the phenyl is rotated nearly perpendicular to the bisecting plane of the cyclohexane ring (Fig. 2b).


When ΔE values were corrected for the difference in zero-point energies between the two conformers, ΔH° values calculated on going from 0 K to higher temperatures, and ΔG° values derived from the calculated entropy differences, the results shown in Table 6 were obtained. The calculated ΔG° at −100 °C, −2.9 kcal / mol, is in excellent accord with the experimental value.

The sizable enthalpy difference between 11-ax and 11-eq (3.1 kcal/mol at 25 °C) can be ascribed to steric repulsion in the axial isomer. On the other hand, the near-zero entropy difference in this equilibrium is in line with the highly based rotameric distribution of 11-ax and 11-eq, adopting the "parallel" and "perpendicular" conformations depicted in figure 2; that is, ΔS° = S°(11-ax) − S°(11-eq) ≈ R ln 2 − R ln 2 ≈ 0.
Thermodynamics of the axial  equatorial conformational equilibrium of tert-butylcyclohexane [14]
equatorial conformational equilibrium of tert-butylcyclohexane [14]
In contrast with methyl-, ethyl-, and isopropylcyclohexane, the axial isomer of tert-butylcyclohexane necessarily orients a methyl group inside the ring, giving rise to a large steric repulsion in axial tert-butylcyclohexane (12-ax), equation 17.

The extreme one-sidedness of equilibrium 17 has precluded so far the experimental estimation of the enthalpic and entropic contributions to −ΔG°(t-Bu) [15]. While this situation brings to mind the potential of theoretical calculations as an alternative for determinations not amenable to experiment, apparently only one force-field study [16] has addressed the question of entropy difference in the 12-ax  12-eq equilibrium (Eq. 17). The estimated ΔS° = 0 for this equilibrium seems intuitively plausible by consideration of the three isoenergetic staggered conformers both in 12-ax and 12-eq.
12-eq equilibrium (Eq. 17). The estimated ΔS° = 0 for this equilibrium seems intuitively plausible by consideration of the three isoenergetic staggered conformers both in 12-ax and 12-eq.
The fundamental importance of the tert-butyl group in chemistry motivated the reexamination of the enthalpic and entropic contributions to the conformational preference of tert-butyl in cyclohexane [14]. The MM2 [17] and MM3 [18] force fields were used to evaluate the intramolecular energetics. While the former program has proven quite successful for modeling a large variety of hydrocarbons, MM3 does take into account entropy components to free energy.
The intramolecular entropy was calculated according to equation 18 where R is the gas constant, n is the number of conformational states sampled, and Pi is the Boltzmann probability of the ith conformational state [19]. The Pi, in turn, were computed from the relationship depicted in equation 19.

Figure 3 presents the MM2 energy profiles for rotation around the C-C(CH3)3 bond in axial and equatorial 12. The most interesting feature of these plots is the presence of two minima for each staggered arrangement in axial 12, relative to only one for each staggered rotamer in 12-eq.

Figure 4 clearly shows that a libration phenomenom results in twice as many conformational states available to axial tert-butylcyclohexane relative to the equatorial isomer.

This is reflected in increased entropy content for 12-ax, as confirmed in the calculated ΔS°ax/eq = S°a x − S°eq = −0.44 cal / degree · mol [14].
The calculated difference in enthalpy between axial and equatorial 12, ΔH°ax/eq = −5.0 kcal/mol, agrees quite well with the value determined by Eliel, ΔG°ax/eq = − 4.9 kcal/mol [20, 21].
Conclusion
Variable-temperature NMR spectroscopy and theoretical calculations are powerful tools for the determination of thermodynamic parameters. It is clear that a proper understanding of the conformational behavior of substituted cyclohexanes requieres knowledge of the enthalpic and entropic components to the conformational free energy difference.
References and notes
1. Juaristi, E. Introduction to Stereochemistry and Conformational Analysis; Wiley: New York, 1991. [ Links ]
2. Booth, H.; Everett, J. R. J. Chem. Soc., Perkin Trans. 2, 1980, 255-259. [ Links ]
3. Juaristi, E.; Labastida, V.; Antúnez, S. J. Org. Chem. 1991, 56, 4802-4804. [ Links ]
4. The methyl group in cis-1 serves as "counterpoise", so that the conformational equilibrium is not overly displaced toward the form with equatorial benzyl [1].
5. Eliel, E. L. Chem Ind. (Londres) 1959, 568-570. [ Links ]
6. Note that temperature is given in Kelvin degrees.
7. Juaristi, E.; Labastida, V.; Antúnez, S. J. Org. Chem. 2000, 65, 969-973. [ Links ]
8. The values reported in Table 3 have been "corrected" for the presence of the counterpoise substituent, the methyl group.
9. Juaristi, E. "Fisicoquímica Orgánica"; Minal: México, 1998. [ Links ]
10. Wiberg, K. B.; Hammer. J. D.; Castejon, H.; Bailey, W. F.; DeLeon, E. L.; Jarrett, R. M. J. Org. Chem. 1999, 64, 2085-2095. [ Links ]
11. Schneider, H. J.; Freitag, W. J. Am. Chem. Soc. 1976, 98, 478-481. [ Links ]
12. Wiberg, K. B.; Castejon, H.; Bailey, W. F.; Ochterski, J. J. Org. Chem. 2000, 65, 1181-1187. [ Links ]
13. Eliel, E. L.; Manoharan, M. J. Org. Chem. 1981, 46, 1959-1962. [ Links ]
14. Antúnez, S.; Juaristi, E. J. Org. Chem. 1996, 61, 6465-6469. [ Links ]
15. Furthermore, an axial tert-butyl group usually causes the ring to adopt non-chair conformations: Remijnse, J. D.; Bekkum, H. V.; Wepster, B. M. Recl. Trav. Chim. Pays-Bas, 1974, 93, 93-98. [ Links ]
16. Allinger, N. L.; Hirsch, J. A.; Miller, M. A.; Tyminski, I. J.; Van Catledge, F. A. J. Am. Chem. Soc. 1968, 90, 1199-1210. [ Links ]
17. Allinger, N. L. J. Am. Chem. Soc. 1977, 99, 8127-8134. [ Links ]
18. Allinger, N. L.; Yuh, Y. H.; Lii, J.-H. J. Am. Chem. Soc. 1989, 111, 8551-8566. [ Links ]
19. Flory, P. J. Statistical Mechanics of Chain Molecules; Wiley: New York, 1969. [ Links ]
20. Manoharan, M.; Eliel, E. L. Tetrahedron Lett. 1984, 25, 3267-3268. [ Links ]
21. See, also: Freeman, F.; Tsegai, Z. M.; Kasmer, M. L.; Hehre, W. J. J. Chem. Educ. 2000, 77, 661-667. [ Links ]

