










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de la Sociedad Química de México
Print version ISSN 0583-7693
Rev. Soc. Quím. Méx vol.45 n.4 Ciudad de México Oct./Dec. 2001
Investigación
Hindered Rotation in N-Carbomethoxylated Indole Derivatives
Martha S. Morales-Ríos, Norma F. Santos-Sánchez and Pedro Joseph-Nathan*
Departamento de Química, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, Apartado 14-740 México 07000, D. F. México. Phone: (52-55) 5747-7112; Fax: (52-55) 5747-7137. E-mail: pjoseph@nathan.chem.cinvestav.mx
Recibido el 25 de octubre del 2001.
Aceptado el 7 de diciembre del 2001.
Dedicated to Dr. Fernando Walls in occasion of its seventy anniversary.
Abstract
N-Carbomethoxyindole derivatives display 1H NMR dynamic processes arising from the hindered rotation about the N-C carbamate bond. Theoretical modeling by Molecular Mechanics predicts two conformational minima A and B, owing to the E and Z isomers of the carbamate group. Conformer A, with the carbonyl carbamate group oriented towards the benzene ring is, in most cases, more stable than the B conformer. A hydrogen bond provides an explanation for the preference of the B conformer in 2. The energy profiles reveal that, between the molecules investigated, compound 3 has the lowest barrier to rotation for the interconversion of the A and B conformers. The relative energies of 3 favored the A conformer in large proportion (97:3). These results are consistent with the observed sharp signals in the 1H NMR spectrum of 3, while compounds 2, 6 and 7 show some significantly broadened signals in their 1H and 13C NMR spectra.
Keywords: N-carbomethoxyindoles, hindered rotation, dynamic processes, NMR.
Resumen
Los derivados de indoles N-carbometoxilados exhiben procesos dinámicos en RMN 1H resultantes de la rotación restringida alrededor del enlace N-C del carbamato. El modelado teórico por mecánica molecular predice dos mínimos conformacionales A y B, debido a los isómeros E y Z del grupo carbamato. El confórmero A, que presenta al carbonilo del carbamato orientado hacia el anillo de benceno es, en la mayoría de los casos, más estable que el confórmero B. La preferencia del confórmero B en 2 se atribuye a la presencia de un puente de hidrógeno. Los perfiles energéticos indican que, entre las moléculas investigadas, el compuesto 3 tiene la menor barrera rotacional para la interconversión de los confórmeros A y B. Las energías relativas de 3 favorecen al confórmero A en amplia proporción (97:3). Estos resultados son consistentes con las señales agudas observadas en el espectro de RMN 1H de 3, mientras que los compuestos 2, 6 y 7 muestran algunas señales significativamente anchas en sus espectros de RMN de 1H y 13C.
Palabras clave: indoles N-carbometoxilados, rotación restringida, procesos dinámicos, RMN.
Introduction
Dynamic NMR studies about the barriers to internal rotation related to amide bonds have shown E and Z conformers in N-formylindoles [1]. However, for N-acetylindole derivatives a single favored conformer (more than 90 %) has been observed, even at low temperatures [2]. In these cases, the carbonyl group was preferentially oriented towards the benzene ring, with the H-7 proton laying in the deshielding current, provided steric or hydrogen bonding effects are not operative [2, 3]. On the other hand, the existence of equilibrating rotamers due to the carbomethoxyl substituent on the indole nitrogen has been well documented [4].
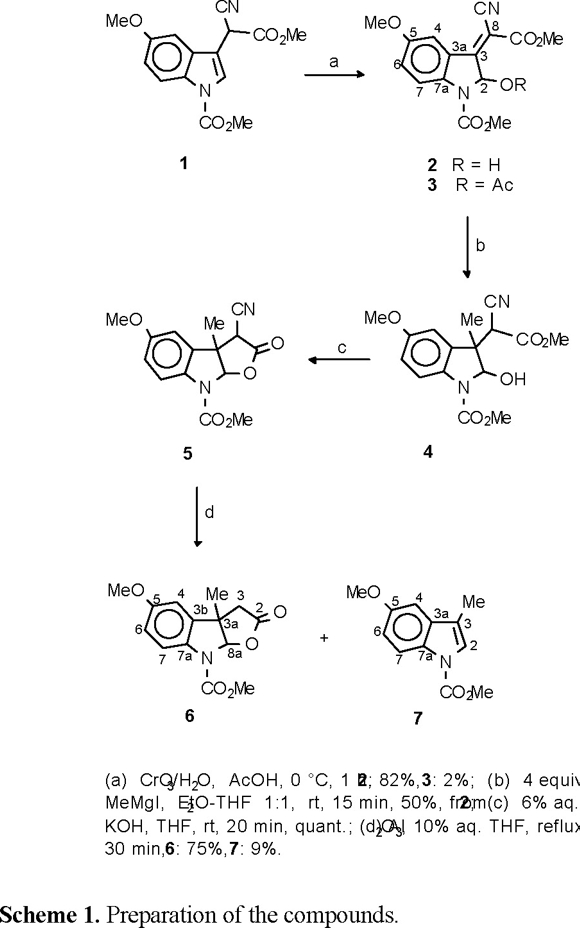
As part of an ongoing study of N-carbomethoxyindole derivatives, as intermediates for the total synthesis of bioactive physostigmine type alkaloids [5], as outlined in Scheme 1, we became interested in exploring which structural changes could influence the hindered rotation that some of these compounds evidenced in their 1H and 13C NMR spectra [6]. For these purposes the N-carbomethoxylated indole derivatives 2, 3, 6 and 7 were investigated.
Results and discussion
Compounds 2, 6 and 7 were found to display dynamic effects in their 1H and 13C NMR spectra. The proton NMR spectrum of 2, measured at room temperature in DMSO-d6, showed broad H-7 (7.75 ppm) and carbamate methyl (3.79 ppm) signals, whereas H-2 bonded to an sp3 hybridized carbon atom remained sharp. On heating, both H-7 and the carbamate methyl signals became sharp, indicating that the rotational barrier was exceeded at the C-N carbamate linkage. Theoretical data (Molecular Mechanics [7]) predict that conformers A and B (Fig. 1) are within an energy range of only 0.4 kcal mol−1 in favor of B (Table 1). The formation of a weak hydrogen bond between the hydroxyl group, attached to an sp3 hybridized C-2 atom, and the carbonyl oxygen of the carbamate group could be at the origin of the slightly more stable B conformation. The hydrogen-oxygen distance was calculated to be 2.2 Å (Fig. 1), which is in the range for intramolecular hydrogen bonding.
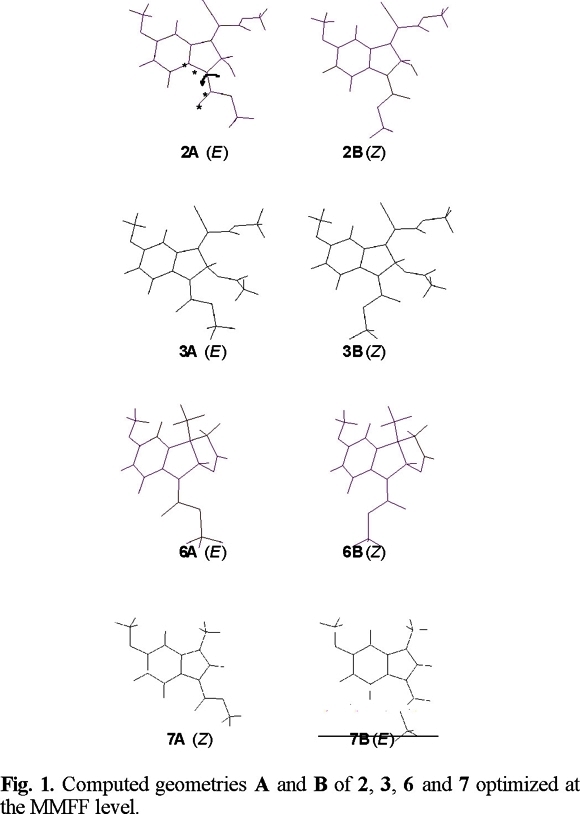
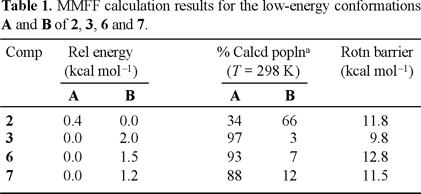
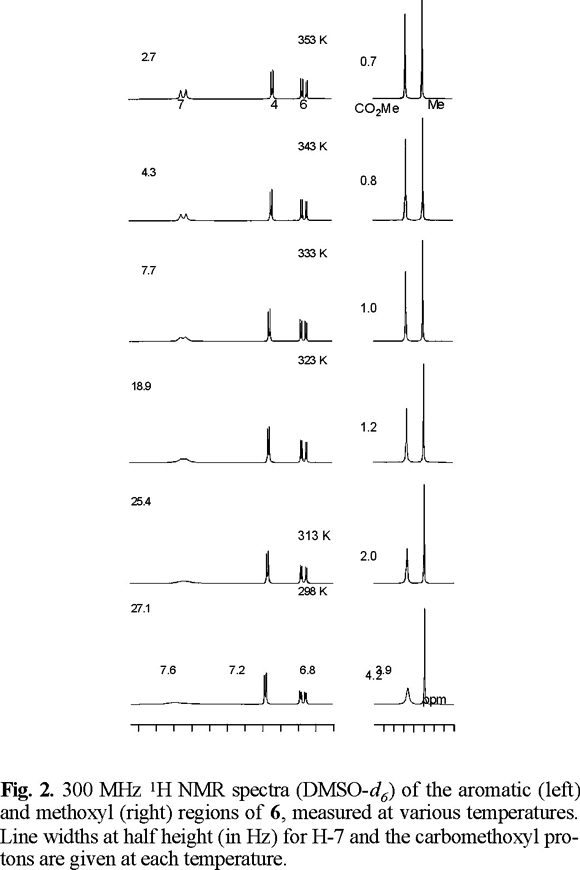
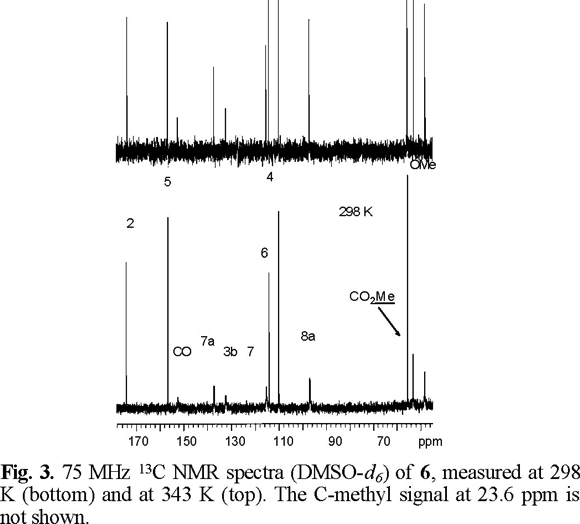
In the variable temperature 1H NMR spectra of 6 measured in DMSO-d6, the signals of H-7 (7.60 ppm) and the carbamate methyl (3.83 ppm) group are significantly broadened at temperatures between 298 and 333 K. Clear sharpening was observed for these resonances above 343 K. At 353 K the signal owing to H-7 appears as doublet with Jortho = 8.8 Hz (Fig. 2). The 13C NMR spectrum of 6, measured at room temperature, showed considerable broadening for the resonances owing to the two carbons of the N-carbomethoxyl group (152.4 and 53.2 ppm), C-7a (137.2 ppm), C-3b (132.3 ppm) C-7 (115.2 ppm), C-8a (96.7 ppm) and C-3a (48.3 ppm), which sharpened at 343 K (Fig. 3). For compound 6, theoretical calculations predict that conformation A (Fig. 1), with the carbonyl carbamate group oriented towards the benzene ring, is preferred over B in a 13:1 ratio and with a relative energy difference of 1.5 kcal mol−1 (Table 1). The lower stability of B appears to result from the electrostatic repulsion between the electron lone pairs of the oxygen C=O carbamate group and those of the oxygen furan ring.
Examination of the proton NMR spectrum of 7, measured at ambient temperature (298 K) in CDCl3, reveals that the signals due to H-7 (8.00 ppm), H-2 (7.32 ppm) and the carbamate methyl (3.98 ppm) group are very broad. When measurements were carried out in DMSO-d6 the signals were significantly sharpened, suggesting a fast rotation at the C-N carbamate linkage on the NMR time scale. For compound 7 theoretical calculations predict two conformational minima, A and B (Fig. 1). Conformation A with the carbonyl carbamate group oriented towards the benzene ring is preferred over B by a relative energy difference of 1.2 kcal mol−1, which corresponds to the 7:1 ratio (Table 1).
The proton NMR spectrum of 3, measured at room temperature in both CDCl3 and DMSO-d6, showed sharp signals for all resonances, which fits with fast interconverting conformers on the NMR time scale. For compound 3, calculations for both conformational minima, A and B, suggest that the one with E arrangement is the most stable by 2.0 kcal mol−1.
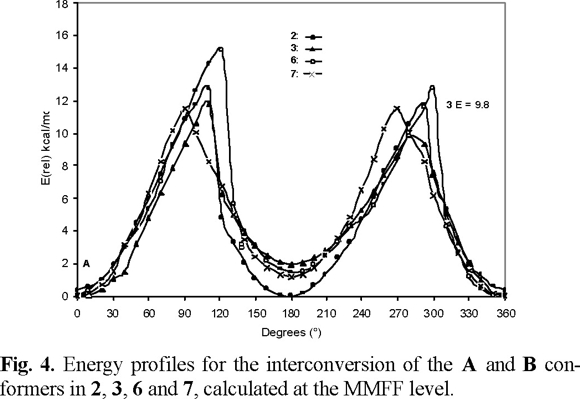
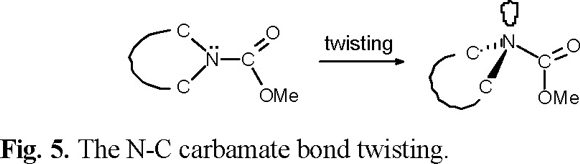
The dihedral drive option in the Spartan Pro program allowed us to calculate barriers to rotation of the carbamate group in 2, 3, 6 and 7, the corresponding data are collected in Table 1. As shown in figure 4, the two ground state conformations A and B of compounds 2, 3, 6 and 7, can be converted into each other by overcoming energy barriers of 11.8, 9.8, 12.8 and 11.5 kcal mol−1, respectively. These barriers are smaller than the activation barriers determined for E-Z isomerisation in amides, which lies in the 14.6-21.5 kcal mol−1 range [8]. As in amides, the carbamate group prefers to be planar (A or B conformations). Experimental evidence for this lies in crystal structure determinations carried out in N-carbomethoxylated indoles [9]. Twisting is accompanied by pyramidalisation at nitrogen as n-ϖ*C=O_ delocalization is progressively turned off, until it disappears completely at a twist of ca. 90° (270°), where the lone pair lies in the nodal plane of the ϖ-system of the carbonyl group (Fig 5). The 90° (270°) rotamer represents a higher energy conformation. For compound 3, the corresponding computed barrier to rotation is the lowest, by ca. 2 to 3 kcal mol−1, allowing the molecule to exists in a fast equilibrium of conformers A and B, with the weighted average strongly in favor of A. These results are in good agreement with the observed sharp signals in the 1H NMR spectrum of 3, measured at 298 K.
Experimental
Melting points were obtained on a Fisher-Johns melting point apparatus and are uncorrected. IR spectra were obtained using a Perkin Elmer 16F PC FT-IR spectrophotometer. EIMS were obtained on Varian Saturn 2000 or Hewlett Packard 5989A spectrometers. NMR spectra were recorded on Varian Mercury spectrometers working at 300 and 75.4 MHz for 1H and 13C, respectively. Chemical shifts are reported in ppm down-field from tetramethylsilane. Standard library pulse sequences were used for all NMR experiments. Dynamic NMR measurements were performed with samples thermostated by means of a stream of air heated to required temperatures. Molecular mechanics force field calculations [7] were performed using the PC Spartan Pro program [10]. Compounds 2 and 6 were obtained from 1 as intermediates in the formal total synthesis of the alkaloids physostigmine and physovenine [11], whereas 3 and 7 were obtained as by-products during the same syntheses (Scheme 1).
Methyl (Z)-3-(1-Cyano-2-methoxy-2-oxoethylidene)-2,3-dihydro-2-hydroxy-5-methoxy-1H-indole-1-carboxylate (2) and its O-acetyl derivative 3. To a precooled (0 °C) stirred solution of 1 [11] (3.9 g, 12.9 mmol) in glacial acetic acid (40 mL) was added dropwise a solution of chromium oxide (4.5 g) in water (25 mL). The reaction mixture was stirred for 1 h at 0 °C and then poured onto cracked ice. The crude precipitate, which had formed, was collected by suction filtration and washed with water (3 × 25 mL). The solid residue was dissolved in EtOAc (200 mL), washed with brine, and dried over Na2SO4. The solvent was removed under reduced pressure, and the residue was crystallized from EtOAc to afford 2, as yellow solid (3.37 g, 82 %): mp 173-174 °C (EtOAc / hexane); Rf = 0.26 (4:6, EtOAc / hexane); IR (CHCl3) νmax: 3570, 3030, 2220, 1724 cm−1; 1H NMR (DMSO-d6) δ 7.75 (1H, br s, H-7), 7.71 (1H, d, J = 2.6 Hz, H-4), 7.35 (1H, d, J = 7.9 Hz, OH), 7.27 (1H, dd, J = 9.1, 2.6 Hz, H-6), 6.55 (1H, d, J = 7.9 Hz, H-2), 3.84 (3H, s, O-CH3), 3.79 (3H, br s, O-CH3), 3.75 (3H, s, O-CH3); 13C NMR (DMSO-d6) δ 162.8, 162.2, 155.2, 151.3, 141.3 (br), 124.1, 122.1, 116.0, 115.9, 108.0, 95.4, 82.8, 55.5, 53.0, 52.9 (br); EIMS m/z 318 [M]+ (100), 286 (41), 258 (40).
The filtrate obtained after the isolation of 2 was allowed to stand at room temperature for 5 days. The formed red crystals were filtered to give the O-acetyl derivative 3 (0.11 g, 2 %): mp 145-147 °C (EtOAc / hexane); Rf = 0.43 (4:6, EtOAc / hexane); IR (CHCl3) νmax: 3032, 2218, 1728, 1256 cm−1; 1H NMR (DMSO-d6) δ 7.85 (1H, d, J = 2.7 Hz, H-7), 7.81 (1H, d, J = 2.7 Hz, H-4), 7.59 (1H, s, H-2), 7.35 (1H, dd, J = 9.1, 2.7 Hz, H-6), 3.81 (3H, s, O-CH3), 3.79 (3H, s, O-CH3), 3.77 (3H, s, O-CH3), 1.98 (3H, s, CH3); 13C NMR (DMSO-d6) δ 166.8, 160.9, 158.9, 155.6, 151.3, 141.5, 123.6, 123.2, 116.6, 115.6, 108.2, 97.0, 81.7, 55.7, 53.5, 53.3, 20.5; EIMS m/z 318 [M-42]+ (8), 302 [M-58]+ (63), 243 (100), 199 (49).
Methyl 5-Methoxy-3a-methyl-2-oxo-2,3,3a,8a-tetrahydro-8H-furo[2,3-b]indole-8-carboxylate (6) and its degradation product 7. Compound 5 was obtained from 4 by the procedure described in Ref. [11]. A mixture of 5 (132 mg, 0.44 mmol) and alumina (1.18 g) in 10 % aqueous THF (6 mL) was stirred at reflux until TLC analysis showed complete disappearance of starting material (0.5 h). The alumina was filtered off and washed with EtOAc (5 × 20 mL). The filtrate and the eluates were combined, concentrated, and purified by flash chromatography on silica gel. Elution with increasing concentration of EtOAc in hexane gave indole 7 as pale yellow solid (9 mg, 9 %): mp 49-50 °C (DMSO); Rf = 0.83 (4:6, EtOAc / hexane); IR (CHCl3) νmax: 3012, 1728, 1376, 1266 cm−1; 1H NMR (DMSO-d6) δ 7.93 (1H, br d, J = 8.8 Hz, H-7), 7.41 (1H, m, H-2), 7.06 (1H, d, J = 2.2 Hz, H-4), 6.94 (1H, dd, J = 8.8, 2.2 Hz, H-6), 4.95 (3H, s, O-CH3), 3.82 (3H, s, O-CH3), 2.20 (3H, d, J = 1.3 Hz, CH3); 13C NMR (DMSO-d6) δ 155.6, 150.7, 131.8, 129.3, 123.0, 116.6, 115.2, 112.9, 101.9, 55.4, 53.7, 9.2; EIMS m/z 219 [M]+ (100), 204 (46), 160 (26).
Further elution gave the principal reaction product, furoindole 6 (2:8, EtOAc / hexane), which was isolated as white solid (91 mg, 75 %): mp 136-137 °C (CHCl3 / hexane); Rf = 0.31 (4:6, EtOAc / hexane); IR (CHCl3) νmax: 3018, 1784, 1722, 1212 cm−1; 1H NMR (DMSO-d6) δ 7.77 (1H, br s, H-7), 6.83 (1H, br dd, J = 8.9, 2.3 Hz, H-6), 6.73 (1H, d, J = 2.3 Hz, H-4), 6.12 (1H, br s, H-8a), 3.90 (3H, br s, O-CH3), 3.80 (3H, s, O-CH3), 2.99 (1H, d, J = 17.9 Hz, H-3), 2.84 (1H, d, J = 17.9 Hz, H-3'), 1.43 (3H, s, CH3); 13C NMR (DMSO-d6) δ 173.4, 157.0, 152.6 (br), 136.0 (br), 133.1 (br) 116.2, 114.0, 109.4, 97.5, 55.8, 53.2 (br), 48.1 (br), 41.3, 24.9; EIMS m/z 277 [M]+ (100), 232 (20), 218 (26).
Acknowledgement
This research was supported in part by CONACyT (Mexico).
References
1. (a) Wijnberg, J. B. P. A.; Speckamp, W. N. Tetrahedron, 1978, 34, 2399-2404. [ Links ] (b) Chatterjee A.; Biswas, K. M. J. Org. Chem. 1973, 38, 4002-4004. [ Links ] (c) Buchardt, O.; Kumler, P. L.; Lohse, C. Acta Chem. Scand. 1969, 23, 1155-1167. [ Links ] (d) Nagarajan, K.; Nair, M. D. Tetrahedron, 1967, 23, 4493-4497. [ Links ] (e) Combrisson, S.; Roques, B. P.; Tetrahedron, 1976, 32, 1507-1516. [ Links ] (f) Garner, G. V.; Meth-Cohn, O.; Suschitzky, H. J. Chem. Soc. (C) 1971, 1234-1236. [ Links ]
2. (a) Monro, A. M.; Sewell, M. J. Tetrahedron Lett. 1969, 7, 595-598. [ Links ] (b) Nagarajan, K.; Nair, M. D.; Pillai, P. M. Tetrahedron, 1967, 32, 1683-1690. [ Links ]
3. Govindachari, T. R.; Pai, B. R.; Rajappa, S.; Viswanathan, N.; Kump, W. G.; Nagarajan, K.; Schmid, H. Helv. Chim. Acta 1962, 45, 1146-1152. [ Links ]
4. (a) Hamburger, M. O.; Cordell, G. A.; Likhitwitayawuid, K.; Ruangrungsi, N. Phytochem. 1988, 27, 2719-2723. [ Links ] (b) Kam. T.-S.; Yoganathan, K.; Li, H.-Y.; Harada, N. Tetrahedron, 1997, 53, 12661-12670. [ Links ] (c) Kam, T.-S.; Yoganathan, K.; Li, H.-Y. Tetrahedron Lett. 1996, 37, 8811-8814. [ Links ]
5. (a) Morales-Ríos, M. S.; Suárez-Castillo, O. R.; Joseph-Nathan, P. J. Org. Chem. 1999, 64, 1086-1087. [ Links ] (b) Morales-Ríos, M. S.; Suárez-Castillo, O. R.; Trujillo-Serrato, J. J.; Joseph-Nathan, P. J. Org. Chem. 2001, 66, 1186-1192. [ Links ] (c) Morales-Ríos, M. S.; Bucio, M. A.; Joseph-Nathan, P. Tetrahedron, 1996, 52, 5339-5348. [ Links ]
6. Morales-Ríos, M. S.; Joseph-Nathan, P. Magn. Reson. Chem. 1987, 25, 911-918. [ Links ]
7. Burket, U.; Allinger, N. L. Molecular Mechanics; ACS Monograph 177, American Chemical Society: Washington, D. C. 1982. [ Links ]
8. Eliel, E. L.; Wilen, S. H. In: Stereochemistry of Carbon Compounds, Wiley-Interscience, New York, 1994, pp. 23, 554. [ Links ]
9. (a) Morales-Ríos, M. S.; Suárez-Castillo, O. R.; Joseph-Nathan, P. J. Chem. Soc., Perkin Trans 2, 2000, 769-775. [ Links ] (b) Morales-Ríos, M. S.; Suárez-Castillo, O. R.; García-Martínez, C; Joseph-Nathan, P. Synthesis, 1998, 1755-1759. [ Links ] (c) Morales-Ríos, M. S.; Suárez-Castillo, O. R.; Alvarez-Cisneros, C.; Joseph-Nathan, P. Can. J. Chem. 1999, 77, 130-137. [ Links ] (d) Morales-Ríos, M. S.; Bucio-Vásquez, M. A.; Joseph-Nathan, P. J. Heterocyc. Chem. 1993, 30, 953-956. [ Links ]
10. Hehre, W. J.; Deppmeier, B. J.; Klunzinger, P. E. A PC Spartan Pro Tutorial, Wavefunction, Inc., Irvine CA, 1999. [ Links ]
11. Morales-Ríos, M. S.; Santos-Sánchez, N. F.; Joseph-Nathan P. J. Nat. Prod. 2002, in press.

